Study scientists identified several currently available drugs that could inhibit growth of these “streaming filaments,” which infected cells use to go after non-infected cells
Like a scene from a bad horror movie, scientists have discovered that SARS-CoV-2, the coronavirus responsible for the COVID-19 pandemic, may be even more sinister and macabre than previously thought. The new research findings will interest those pathologists and clinical laboratory professionals who want to understand how the coronavirus spreads once it enters the body.
Headed by scientists from the University of San Francisco (UCSF), a team of international researchers discovered that “when the SARS-CoV-2 virus infects a human cell, it sets off a ghoulish transformation,” reported the Los Angeles Times (LA Times).
“Obeying instructions from the virus,” the LA Times continued, “the newly infected cell sprouts multi-pronged tentacles studded with viral particles. These disfigured zombie cells appear to be using those streaming filaments, or filopodia, to reach still-healthy neighboring cells. The protuberances appear to bore into the cells’ bodies and inject their viral venom directly into those cells’ genetic command centers—thus creating another zombie.”
As If the Coronavirus Weren’t Bad Enough!
“It’s just so sinister that the virus uses other mechanisms to infect other cells before it kills the cell,” Nevan Krogan, PhD, Professor, Department of Cellular Molecular Pharmacology at the UCSF School of Medicine, one of the study’s authors, told the LA Times.
The images above taken with an electron microscope show the streaming filaments—or as the researchers described in their published study, “filopodial protrusions possessing budding viral particles”—reaching out from cells infected with the SARS-CoV-2 coronavirus looking for other cells to infect. (Photos copyright: Los Angeles Times/Elizabeth Fischer, MA, Chief, RML Microscopy Unit, NIAID/NIH.)
SARS-CoV-2 Has Evolved, Study Suggests
Prior to this discovery, scientists believed that the coronavirus infected cells in a typical fashion by finding receptors on the surface of cells lining an individual’s mouth, nose, respiratory tract, lungs or blood vessels, and eventually replicating and invading larger cells. However, this new research may suggest that the virus has evolved and developed new ways to pass quickly and effectively from cell to cell.
While some other illnesses, including smallpox, human immunodeficiency virus (HIV), and some influenza viruses have been known to use filopodia to enhance their ability to infect cells, Krogan contends that those other viruses do not seem to have the prolific growth of the SARS-CoV-2 filopodia.
“By conducting a systematic analysis of the changes in phosphorylation when SARS-CoV-2 infects a cell, we identified several key factors that will inform not only the next areas of biological study, but also treatments that may be repurposed to treat patients with COVID-19,” he said, in a UCSF news release.
UCSF Study Identifies Drugs, Compounds That May Disrupt Growth of Filopodia
One key finding is that the coronavirus was utilizing a specific type of molecule from a family of cellular helpers known as Kinase to create the filopodia.
The researchers conducted a “quantitative mass spectrometry-based phosphoproteomics survey of SARS-CoV-2 infection in Vero E6 cells,” the study noted, which revealed a “dramatic rewiring of phosphorylation on host and viral proteins.
“SARS-CoV-2 infection promoted casein kinase 2 (CK2) and p38 MAPK activation, production of diverse cytokines, and shutdown of mitotic kinases, resulting in cell cycle arrest,” the study continued, adding, “Infection also stimulated a marked induction of CK2-containing filopodial protrusions possessing budding viral particles.
“Eighty-seven drugs and compounds were identified by mapping global phosphorylation profiles to dysregulated kinases and pathways. We found pharmacologic inhibition of the p38, CK2, CDK, AXL, and PIKFYVE kinases to possess antiviral efficacy, representing potential COVID-19 therapies,” the researchers concluded.
To determine if they might be helpful in combating COVID-19, the UCSF research team tested drugs and compounds that were either already cleared to market by the US federal Food and Drug Administration (FDA), in clinical trials, or under preclinical development.
After discovering the Kinase connection, the scientists focused on specialized drugs known as Kinase inhibitors.
“We narrowed in on about a dozen, and we highlighted about six or seven that look particularly potent in a laboratory setting,” Krogan told ABC News. “And we’re very excited now to try and take these into clinical trials.”
Among the drugs the study identified as potentially being able to disrupt the creation of filopodia and slow the spread of COVID-19 in the body are:
Silmitasertib: A drug that is currently in the clinical trial stages as a treatment for bile duct cancer and other cancers, including hematological and lymphoid malignancies;
“We are encouraged by our findings that drugs targeting differentially phosphorylated proteins inhibited SARS-CoV-2 infection in cell culture,” said Kevan Shokat, PhD, Professor of Cellular and Molecular Pharmacology at UCSF, and co-author of the study, in the UCSF news release. “We expect to build upon this work by testing many other kinase inhibitors, while concurrently conducting experiments with other technologies to identify underlying pathways and additional potential therapeutics that may intervene in COVID-19 effectively.”
Presently, the UCSF study provides no direct benefit to COVID-19 illness patients or clinical laboratories performing SARS-CoV-2 testing. However, that could change rapidly. Pathologists and medical laboratory managers will want to keep an eye on this research, because it may lead to new treatments for COVID-19 that would require increased clinical laboratory testing to identify people infected with the coronavirus.
Though the test initially drew ‘raves’ from Trump administration, the FDA now suggests negative results should be confirmed with an additional ‘high-sensitivity authorized SARS-CoV-2 molecular test’
This spring, as the United States attempted to jump-start a national response to the SARS-CoV-2 coronavirus pandemic, the Trump administration heralded Abbott Laboratories’ five-minute test for COVID-19 as a major breakthrough. But even as the federal Food and Drug Administration (FDA) issued dozens of Emergency Use Authorizations (EUAs) to quickly get COVID-19 diagnostic tests into clinical use, the accuracy of some of those tests came into question—including Abbott’s ID NOW COVID-19 rapid molecular test.
The continuing controversy over Abbott’s ID NOW COVID-19 test shows how the national spotlight can be a double-edged sword, bringing both widespread favorable attention to a breakthrough technology, followed by heightened public scrutiny if deficiencies emerge. At the same time, from the first news stories about the Abbott ID NOW COVID-19 test, pathologists and clinical laboratory managers understood that this test always had certain performance parameters, as is true of every diagnostic test.
“Everybody was raving about it,” a former administration official, speaking on the condition of anonymity to discuss internal deliberations, said of ID NOW in an interview with Kaiser Health News (KHN). “It’s an amazing test, but it has limitations which are now being better understood.”
In a White House ceremony on March 29, 2020, President Trump praised his administration’s role in speeding up development “on both testing and treatment that will help us win our war against the coronavirus.” Among the moves highlighted was the FDA’s approval two days earlier of Abbott’s ID-NOW COVID-19 rapid molecular test (above), which the President stated, “delivers lightning-fast results in as little as five minutes,” adding, “Normally, this approval process from the FDA would take 10 months, and even longer, but we did it in four weeks.” (Photo copyright: Washington Post.)
FDA Warns Public about Inaccurate Test Results
On May 14, the FDA issued a public warning about the point-of-care test’s accuracy after receiving 15 “adverse event reports” indicating some patients were receiving “false negative results.”
“Regardless of method of collection and sample type, Abbott ID NOW COVID-19 had negative results in a third of the samples that tested positive by Cepheid Xpert Xpress when using nasopharyngeal swabs in viral transport media and 45% when using dry nasal swabs,” the NYU study authors stated.
Abbott Rebuts Criticism
In a statement following the FDA’s warning, Abbott said, “We’re seeing studies being conducted to understand the role of ID NOW in ways that it was not designed to be used. In particular, the NYU study results are not consistent with other studies. While we’ve seen a few studies with sensitivity performance percentages in the 80s, we’ve also seen other studies with sensitivity at or above 90%, and one as high as 94%.
“While we understand no test is perfect, test outcomes depend on a number of factors including patient selection, specimen type, collection, handling, storage, transport and conformity to the way the test was designed to be run. ID NOW is intended to be used near the patient with a direct swab test method,” Abbott’s statement added, noting the company would be “further clarifying our product information to provide better guidance” and “reinforcing proper sample collection and handling instructions.”
Then, on May 21, Abbott issued another statement highlighting an interim analysis of an ongoing multisite clinical study demonstrating ID NOW COVID-19 test performance is ≥94.7% in positive agreement (sensitivity) and ≥98.6% negative agreement (specificity) when compared to two different lab-based molecular PCR reference methods.
“We’re pleased ID NOW is delivering on what it was designed to do—quickly detect the virus in people who need to know now if they’re infected,” said Philip Ginsburg, MD, SAIM, Senior Medical Director, Infectious Disease, Rapid Diagnostics at Abbott, in the statement. “This is great news for people who are experiencing symptoms and want to take action before they infect others, reducing the spread of infection in society.”
Nonetheless, KHN reported on June 22 that the FDA had “received a total of 106 reports of adverse events for the Abbott test, a staggering increase. The agency has not received a single adverse event report for any other point-of-care tests meant to diagnose COVID-19.”
Second Comparison Study Results for Abbott’s ID NOW
The Abbott ID NOW test correctly identified 74% of positive samples. In comparison, Cepheid’s Xpert Xpress SARS CoV-2 test correctly identified 99% of positives. Negative agreement was 100% and 92.0% for ID NOW and Xpert, respectively.
The FDA’s testing policy for clinical laboratories and commercial manufacturers recommends diagnostic tests correctly identify at least 95% of positive samples. However, KHN pointed out, a senior FDA official in late May said coronavirus tests that were administered outside lab settings would be considered useful in fighting the pandemic even if they miss 20% of positive cases.
“There’s no way I would be comfortable missing two out of 10 patients,” Whittier told KHN.
Abbott ID-NOW’s Role in the Global Fight to Stop COVID-19
Abbott’s ID NOW COVID-19 test is promoted as delivering positive test results in five minutes and negative results in about 13 minutes. On its website and in news releases, Abbott maintains its test “performs best in patients tested earlier post symptom onset.”
In a July 17 statement, Abbott said, “ We have shipped 5.3 million of our rapid ID NOW tests to all 50 states, Washington DC, Puerto Rico and the Pacific Islands. The majority of these tests have been sent to outbreak hotspots and we’ve asked that customers prioritize frontline healthcare workers and first responders.”
It is common for a new diagnostic instrument and a new clinical laboratory test to be continually improved after initial launch. Thus, the performance of such devices at the time they are given clearance from the FDA to be used in clinical care can be much improved several months or years later.
Given the importance of a reliable point-of-care SARS-CoV-2 test during the pandemic, it can be assumed that Abbott Laboratories is working closely with its medical laboratory customers specifically to improve the accuracy, reliability, and reproducibility of both the instrument and the test kit.
The fledgling test-kit company sent plastic preforms that were intended for use in the manufacturing of soda bottles, not clinical laboratory specimen tubes
When is a specimen tube not a specimen tube? When it is a plastic tube made for creating soda bottles. And that may be exactly what the Federal Emergency Management Agency (FEMA) received after paying $7.3 million to a fledgling Florida-based company that won a multi-million-dollar no-bid contract from the federal government for COVID-19 clinical laboratory testing supplies, which FEMA then shipped nationwide to states that had requested the supplies.
FEMA signed the deal with Fillakit, LLC, on May 7, 2020, “just six days after the company was formed,” reported ProPublica, which went on to state that the shipment of unusable Fillakit specimen tubes contributed to delays in rolling out widespread COVID-19 testing in the US.
According to ProPublica, Fillakit supplied “preforms” that are designed to be expanded with heat and pressure into 2-liter soda bottles, not laboratory specimen tubes.
Michelle Forman, a spokesperson for the Association of Public Health Laboratories, told ProPublica one major flaw of the Fillakit tubes is their size. “They are an unusual shape, so they don’t fit racks,” she said, “and we are getting lots of pushback about how difficult it is to work with them from our clinical partners.”
The photo of the preform sent by Fillakit above is taken from a Fox23 news report that stated “FEMA sent the Washington State Department of Health nearly 300,000 plastic tubes. They thought they were getting test tubes for coronavirus testing, but instead, they received tiny plastic preforms that can be made into 2-liter soda bottles.” This photograph shows the preform tube that is intended to be die-molded into a large soda bottle. That is why the cap on the tube is appropriate for the tubes intended purpose as a soda bottle. (Photo copyright: Alison Grande, KIRO7/Fox23.)
Fillakit Employees Describe ‘Unsanitary’ Working Conditions
Ex-employees of Fillakit told the Wall Street Journal (WSJ) the specimen tubes were being handled in unsanitary open-air conditions in a warehouse outside of Houston where the test kits were being assembled.
“There were up to 250 workers crowded in a small warehouse room, shoulder to shoulder … working off of fold-up tables with supplies placed on the floor and handled without gloves,” Teresa Bosworth Green told Community Impact (CI), which reported that Green worked at Fillakit from May 11-20.
“We were told that we would be filling and capping tubes that would be used for COVID testing,” Green told CI.
However, according to CI, Green “expressed concern about the lack of cleanliness and facemasks. Green brought her own mask, but workers were not initially provided any.”
Green told CI, “People were breathing and coughing right over the solution.”
In a letter to FEMA and the Department of Health and Human Services (HHS) after Michigan received more than 322,000 tubes of transport media manufactured by Fillakit, Democrat Senators Debbie Stabenow and Gary Peters wrote, “Even if the tubes themselves were not unsuitable for testing purposes, the contamination risks inherent in such careless handling would cause serious concerns about the reliability of any tests conducted using these materials.”
On July 7, 2020, the Wall Street Journal reported that Fillakit had notified the Florida Secretary of State on June 26 that the company had been dissolved.
Kira Doyle, JD (above), owner/attorney at Kira Doyle Law in St. Petersburg, Fla., who multiple media outlets listed as Fillakit’s manager, told the Tampa Bay Times that media portraits of the company have been unfair. In a series of emails, she said Fillakit was attempting to fill a void in the medical supply chain. “If you are interested in writing an article about empowered female business owners or entrepreneurs creating jobs and helping this great country during an unprecedented pandemic, Fillakit LLC, fits that profile,” Doyle wrote. (Photo copyright: Kira Doyle Law.)
Under Pressure, Feds Award Contracts for COVID-19 Test Supplies to Inexperienced Suppliers
Fillakit as just one example out of “more than 250 companies that got contracts worth more than $1 million without going through a fully competitive bidding process,” NPR reported.
“Government procurement experts say federal officials were trying to move quickly to deliver desperately needed personal protective equipment,” NPR continued. “But they question the need to turn to contractors who have never worked with the government before and lacked experience making or delivering the protective gear.”
Among those receiving contracts were companies with little to no experience in manufacturing clinical laboratory testing supplies, personal protective equipment (PPE), as well as others that had never worked in the medical field. One company imported vodka, while another was a school security consultant. Many of the contractors served as middlemen, securing PPE from Chinese manufacturers, which meant they often were “competing with federal agencies, state governments, and local health systems,” all of which were attempting to buy the same equipment in the global marketplace, NPR reported.
“Giving business to people who don’t have experience is something you don’t want to do in an emergency,” Joshua Schwartz, JD, a professor of Government Contracts Law and co-director of the Government Procurement Law Program at George Washington University School of Law, told NPR.
FEMA Defends Its Contracting Process
A ProPublica analysis of coronavirus contracts found that about 13% of total federal government pandemic spending went to first-time vendors. And in a follow-up article, ProPublica claimed, “many of the new contractors have no experience acquiring medical products.”
FEMA, however, maintains it pays for purchases only after they have been delivered to minimize potential for waste of taxpayer dollars. “FEMA does not enter into contracts unless it has reason to believe they will be successfully executed,” the agency told ProPublica.
The US’ lack of preparedness for the COVID-19 pandemic has resulted in missteps and misspending as federal agencies struggle to provide hospitals, clinical laboratories, and healthcare providers with personal protective gear and test supplies, and to ramp up COVID-19 testing nationwide.
This is yet another instance where federal agencies appear to lack the competencies required to fulfill healthcare requirements with proven products that meet critical specifications. Meanwhile, in every community throughout the United States, independent medical laboratories and hospital-based laboratories are clamoring for adequate supplies of everything from collect swabs and viral transport media to reagents and cuvettes.
Researchers found evidence indicating that the virus has—under selection pressure—made itself more stable, giving it a “significant boost in infectivity”
While the COVID-19 pandemic continues to spread across the United States and throughout the world, new research suggests that a coming genetic mutation within the SARS-CoV-2 coronavirus may make it much more dangerous than it already is. This finding has significant implications for clinical laboratories that perform COVID-19 testing and the in vitro diagnostics (IVD) companies that develop and manufacture tests for COVID-19.
The mutation, called D614G, will provide the coronavirus with sturdier spikes that will increase its ability to latch onto and infect cells. That’s according to a study conducted at The Scripps Research Institute (Scripps) in Jupiter, Fla., which found that a mutated coronavirus may be up to 10 times more infectious than the original strain.
“Viruses with this mutation were much more infectious than those without the mutation in the cell culture system we used,” said Hyeryun Choe, PhD, Professor, Department of Immunology and Microbiology, Scripps Research, and senior author of the study, in a Scripps news release.
A More Flexible and Potent Coronavirus May Be Coming
The researchers found that coronavirus particles containing the mutation tend to have four to five times more functional spikes than particles without the mutation. The spikes enable the virus to bind to cells more easily. The research suggests that the greater the number of functional spikes on the viral surface the greater the flexibility and potency of the coronavirus.
In the Scripps news release, Farzan said, “more flexible spikes allow newly made viral particles to navigate the journey from producer cell to target cell fully intact, with less tendency to fall apart prematurely.
“Over time, it has figured out how to hold on better and not fall apart until it needs to,” he added. “The virus has, under selection pressure, made itself more stable.”
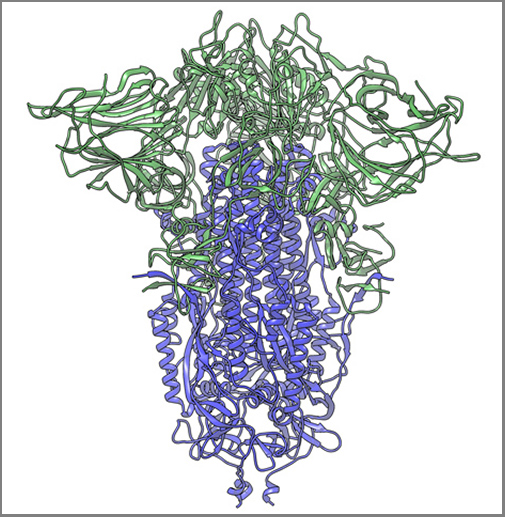
The image above, taken from the Scripps Research news release, shows “a cryogenic electron microscope image of a SARS-CoV-2 spike protein side view, the S1 section of the spike is shown in green and the S2 portion is shown in purple. This unique two-piece system has shown itself to be relatively unstable. A new mutation has appeared in the viral variant most common in New York and Italy that makes this spike both more stable and better able to infect cells.” (Graphic and caption copyright: Andrew Ward lab, Scripps Research.)
Mutation Makes SARS-CoV-2 Coronavirus ‘Much More Stable’
The two Scripps scientists have studied coronaviruses for nearly 20 years and performed extensive research on the Severe Acute Respiratory Syndrome (SARS) outbreak that occurred in 2003. They noted that there is a difference between spike proteins of SARS, an earlier strain of coronavirus, and the new SARS-CoV-2 strain.
The protein spikes of both strains were originally tripod shaped. However, the spikes of the SARS-CoV-2 coronavirus are divided into two different segments: S1 and S2. According to the published study: “The S1domain mediates receptor binding, and the S2 mediates downstream membrane fusion.”
This feature originally produced unstable spikes, but with the D614G mutation, the tripod breaks less frequently, which makes more of the spikes fully functional and the virus more infectious.
“Our data are very clear, the virus becomes much more stable with the mutation,” Choe said in the news release.
Is COVID-19 Spread Due to ‘Founder Effect’
The scientists also examined whether the spread of COVID-19 could have been the result of the “Founder Effect,” which is seen when a small number of variants fan out into a wide population by chance. Could the founder effect explain why COVID-19 outbreaks in some areas of the world were more severe than others? The researchers believe their data definitively answered that question.
“There have been at least a dozen scientific papers talking about the predominance of this mutation,” Farzan said. “Are we just seeing a founder effect? Our data nails it. It is not the founder effect.”
Hyeryun Choe, PhD (left), and Michael Farzan, PhD (right), scientists at Scripps Research explained that their research was performed using engineered viruses and that their observations of the virus and its mutation may not translate to increased transmissibility when a virus attaches to a host outside the lab. COVID-19 and its mutation appear to be relatively stable and are mutating at a rate slower than that of the seasonal flu, which may be critical factors in the development of a vaccine. (Photos copyright: Scripps Research.)
Findings Raise ‘Interesting’ Questions about the COVID-19 Coronavirus
Nevertheless, the two scientists are curious about some of their findings. “Our data raise interesting questions about the natural history of SARS-CoV-2 as it moved presumably from horseshoe bats to humans. At some point in this process, the virus acquired a furin-cleavage site, allowing its S1/S2 boundary to be cleaved in virus-producing cells. In contrast, the S1/S2 boundary of SARS-CoV-1, and indeed all SARS-like viruses isolated from bats, lack this polybasic site and are cleaved by TMPRSS2 or endosomalcathepsins in the target cells.
“In summary, we show that an S protein mutation that results in more transmissible SARS-CoV-2 also limits shedding of the S1 domain and increases S-protein incorporation into the virion. Further studies will be necessary to determine the impact of this change on the nature and severity of COVID-19,” the Scripps researchers concluded.
However, not all Scripps researchers agreed with the conclusions of Choe and Farzan’s research.
The Times of Israel reported that Kristian Andersen, PhD, a professor in the Department of Immunology and Microbiology, Scripps California Campus, told the New York Times that “other analyses of virus variants in labs had not found significant differences in infection rates.”
“That’s the main reason that I’m so hesitant at the moment,” Andersen said. “Because if one really was able to spread significantly better than the other, then we would expect to see a difference here, and we don’t.”
Times of Israel also reported that “In late May researchers in University College London said their studies of the genomes of more than 15,000 samples had not shown one strain being more infectious than others.”
So, the jury’s out. Nonetheless, clinical laboratory leaders will want to remain vigilant. A sudden increase in COVID-19 infection rates will put severe strain on already strained laboratory supply chains.
As digital healthcare continues to gain acceptance and regulatory support, clinical laboratories will need to help patients provide biological samples for virtual doctor visits
Patterns are emerging in healthcare that will likely impact clinical laboratories now and into the future. Trends in telehealth and mobile health (mHealth) that were just beginning to develop before the COVID-19 pandemic have accelerated with the outbreak, and many are predicted to remain once the pandemic is over, reported Healthcare Business and Technology.
Now comes virtual waiting rooms to go along with virtual doctor’s visits. One example is Banner Health of Phoenix, Arizona. The non-profit has more than 50,000 employees in Ariz. and is the state’s largest employer. It operates 28 hospitals and multiple specialty clinics in six states, making it one of the largest health systems in the US as well.
Banner Health is working with LifeLink to deploy virtual waiting rooms in all of its 300 clinics.
What is a Virtual Waiting Room?
The Banner Health System includes 1,500 physicians who work in 300 clinics. More than one million patients in Arizona, California, Colorado, Nebraska, Nevada, and Wyoming are part of the system.
In the not too distant past, when patients visited Banner Health providers and received doctor’s orders for diagnostic tests, they then went to clinical laboratories or the lab’s patient service centers to provide a biological specimen for testing.
Now, because of COVID-19, patients at Banner Health clinics access virtual waiting rooms through a mobile device or computer. They check in virtually for video visits and may not visit a doctor’s office or medical facility at all. Instead, they engage their healthcare provider through a telehealth connection.
The introduction of the virtual waiting rooms is Banner Health’s response to the need for social distancing during the COVID-19 pandemic.
The virtual waiting rooms employ LifeLink chatbots, which interact with patients in a conversational way, and are available for both telehealth and in-person appointments. The chatbots can:
provide appointment reminders,
guide patients through completing necessary paperwork,
provide instructions on using telehealth technology,
check patients in for appointments, and
direct patients to an exam room for in-person doctor visits.
Banner Health used similar technology for patients visiting their emergency departments.
“The traditional patient experience of walking into an office, filling out paper forms, reading instructions and then waiting for an exam room had to change. LifeLink chatbots have already helped hundreds of thousands of Banner patients navigate emergency room visits, so the concept of digitizing regular doctor appointment visits with a mobile, virtual waiting room chatbot assistant was a natural extension of the technology,” said Jeff Johnson, JD, Vice President of Innovation and Digital Business at Banner Health, in a press release. (Photo copyright: Healthcare IT News.)
Both Patients and Healthcare Providers Need to Adapt
“The COVID-19 pandemic requires an entirely different level of thinking when it comes to providing routine patient services,” said Greg Johnsen, CEO at LifeLink, in the Banner Health press release. “Like the changes we are seeing in retail, healthcare providers need to adapt, and the waiting room experience is one area that will need to change. We take great pride in knowing that LifeLink chatbots are providing peace of mind and convenience for patients that need to see their doctors.”
A significant innovation is that patients can easily engage with the chatbots through a “one-click authentication process and then interact through a standard web browser,” rather than requiring them to download and install a mobile device app, Healthcare IT News reported.
“One of the key benefits of this chatbot technology is the ease of use,” said Banner Health’s Jeff Johnson in the press release. “Interactions that use natural language eliminate the need for user training, and there are no apps or passwords required so it’s simple for patients to interact with us securely, on any device. We have seen high engagement rates as a result.”
One thing seems certain, as COVID-19 causes increased anxiety over social distancing, it is likely that virtual healthcare, telehealth, and digital pathology will continue to be developed in the medical industry.
This has implications for clinical laboratories, because if patients are being scheduled virtually, it is just a small additional step to have the doctor see them virtually via telehealth. In such circumstances, medical laboratories will need to have a way for the patient to provide a specimen for lab testing.
In the absence of a “gold standard,” researchers are finding a high frequency of false negatives among SARS-CoV-2 RT-PCR tests
Serology tests designed to detect antibodies to the SARS-CoV-2 coronavirus that causes the COVID-19 illness have been dogged by well-publicized questions about accuracy. However, researchers also are raising concerns about the accuracy of molecular diagnostics which claim to detect the actual presence of the coronavirus itself.
“Diagnostic tests, typically involving a nasopharyngeal swab, can be inaccurate in two ways,” said Steven Woloshin, MD, MS, in a news release announcing a new report that “examines challenges and implications of false-negative COVID-19 tests.” Woloshin is an internist, a professor at Dartmouth Institute, and co-director of the Geisel School of Medicine at Dartmouth.
“A false-positive result mistakenly labels a person infected, with consequences including unnecessary quarantine and contact tracing,” he stated in the news release. “False-negative results are far more consequential, because infected persons who might be asymptomatic may not be isolated and can infect others.”
Woloshin led a team of Dartmouth researchers who analyzed two studies from Wuhan, China, and a literature review by researchers in Europe and South America that indicated diagnostic tests for COVID-19 are frequently generating false negatives. The team published their results in the June 5 New England Journal of Medicine (NEJM).
For example, one research team in Wuhan collected samples from 213 hospitalized COVID-19 patients and found that an approved RT-PCR test produced false negatives in 11% of sputum samples, 27% of nasal samples, and 40% of throat samples. Their research was published on the medRxiv preprint server and has not been peer-reviewed.
The literature review Woloshin’s team studied was also published on medRxiv, titled, “False-Negative Results of Initial Rt-PCR Assays for COVID-19: A Systematic Review.” It indicated that the rate of false negatives could be as high as 29%. The authors of the review looked at five studies that had enrolled a total of 957 patients. “The collected evidence has several limitations, including risk of bias issues, high heterogeneity, and concerns about its applicability,” they wrote. “Nonetheless, our findings reinforce the need for repeated testing in patients with suspicion of SARS-Cov-2 infection.”
Another literature review, published in the Annals of Internal Medicine, titled, “Variation in False-Negative Rate of Reverse Transcriptase Polymerase Chain Reaction–Based SARS-CoV-2 Tests by Time Since Exposure,” estimated the probability of false negatives in RT-PCR tests at varying intervals from the time of exposure and symptom onset. For example, the authors found that the median false-negative rate was 38% if a test was performed on the day of symptom onset, versus 20% three days after onset. Their analysis was based on seven studies, five of which were peer-reviewed, with a total of 1330 test samples.
Doctors also are seeing anecdotal evidence of false negatives. For example, clinicians at UC San Diego Health medical center treated a patient with obvious symptoms of COVID-19, but two tests performed on throat samples were negative. However, a third test, using a sample from a bronchial wash, identified the virus, reported Medscape.
Sensitivity and Specificity of COVID-19 Clinical Laboratory Tests
The key measures of test accuracy are sensitivity, which refers to the ability to detect the presence of the virus, and specificity, the ability to determine that the targeted pathogen is not present. “So, a sensitive test is less likely to provide a false-negative result and a specific test is less likely to provide a false-positive result,” wrote Kirsten Meek, PhD, medical writer and editor, in an article for ARUP Laboratories.
“Analytic” sensitivity and specificity “represent the accuracy of a test under ideal conditions in which specimens have been collected from patients with either high viral loads or a complete absence of exposure,” she wrote. However, “sensitivity and specificity under real-world conditions, in which patients are more variable and specimen collection may not be ideal, can often be lower than reported numbers.”
In a statement defending its ID Now molecular point-of-care test, which came under scrutiny during a study of COVID-19 molecular tests by NYU Langone Health, Northwell Health, and Cleveland Clinic, according to MedTech Dive, Abbott Laboratories blamed improper sample collection and handling for highly-publicized false negatives produced by its rapid test. An FDA issued alert about the test on May 14 noted that Abbott had agreed to conduct post-market studies to identify the cause of the false negatives and suggest remedial actions.
Issues with Emergency Use Authorizations
In their NEJM analysis, Woloshin et al point to issues with the FDA’s process for issuing Emergency Use Authorizations (EUAs). For example, they noted variations in how manufacturers are conducting clinical evaluations to determine test performance. “The FDA prefers the use of ‘natural clinical specimens’ but has permitted the use of ‘contrived specimens’ produced by adding viral RNA or inactivated virus to leftover clinical material,” they wrote.
When evaluating clinical performance, manufacturers ordinarily conduct an index test of patients and compare the results with reference-standard test, according to the Dartmouth researchers. For people showing symptoms, the reference standard should be a clinical diagnosis performed by an independent adjudication panel. However, they wrote, “it is unclear whether the sensitivity of any FDA-authorized commercial test has been assessed in this way.” Additionally, a reference standard for determining sensitivity in asymptomatic people “is an unsolved problem that needs urgent attention to increase confidence in test results for contact-tracing or screening purposes.”
“To truly determine false negatives, you need a gold standard test, which is essentially as close to perfect as we can get,” Stephen Rawlings, MD, PhD, (above), a resident physician of internal medicine and infectious diseases fellow at UC San Diego’s Center for AIDS Research (CFAR), who has been working on SARS-CoV-2 test validation since March. “But there just isn’t one yet for coronavirus,” he told Medscape. (Photo copyright: University of California, San Diego.)
Continued adherence to current measures, such as physical distancing and surface disinfection.
Development of highly sensitive and specific tests or combinations of tests to minimize the risk of false-negative results and ongoing transmission based on a false sense of security.
Improved RT-PCR tests and serological assays.
Development and communication of clear risk-stratified protocols for management of negative COVID-19 test results.
“These protocols must evolve as diagnostic test, transmission, and outcome statistics become more available,” they wrote.
Meanwhile, clinical laboratories remain somewhat on their own at selecting which COVID-19 molecular and serology tests they want to purchase and run in their labs. Complicating such decisions is the fact that many of the nation’s most reputable in vitro diagnostics manufacturers cannot produce enough of their COVID-19 tests to meet demand.
Consequently, when looking to purchase tests for SARS-CoV-2, smaller medical laboratory organizations find themselves evaluating COVID-19 kits developed by little-known or even brand-new companies.