Researchers intend their new AI image retrieval tool to help pathologists locate similar case images to reference for diagnostics, research, and education
Researchers at Stanford University turned to an unusual source—the X social media platform (formerly known as Twitter)—to train an artificial intelligence (AI) system that can look at clinical laboratory pathology images and then retrieve similar images from a database. This is an indication that pathologists are increasingly collecting and storing images of representative cases in their social media accounts. They then consult those libraries when working on new cases that have unusual or unfamiliar features.
The Stanford Medicine scientists trained their AI system—known as Pathology Language and Image Pretraining (PLIP)—on the OpenPath pathology dataset, which contains more than 200,000 images paired with natural language descriptions. The researchers collected most of the data by retrieving tweets in which pathologists posted images accompanied by comments.
“It might be surprising to some folks that there is actually a lot of high-quality medical knowledge that is shared on Twitter,” said researcher James Zou, PhD, Assistant Professor of Biomedical Data Science and senior author of the study, in a Stanford Medicine SCOPE blog post, which added that “the social media platform has become a popular forum for pathologists to share interesting images—so much so that the community has widely adopted a set of 32 hashtags to identify subspecialties.”
“It’s a very active community, which is why we were able to curate hundreds of thousands of these high-quality pathology discussions from Twitter,” Zou said.
“The main application is to help human pathologists look for similar cases to reference,” James Zou, PhD (above), Assistant Professor of Biomedical Data Science, senior author of the study, and his colleagues wrote in Nature Medicine. “Our approach demonstrates that publicly shared medical information is a tremendous resource that can be harnessed to develop medical artificial intelligence for enhancing diagnosis, knowledge sharing, and education.” Leveraging pathologists’ use of social media to store case images for future reference has worked out well for the Stanford Medicine study. (Photo copyright: Stanford University.)
Retrieving Pathology Images from Tweets
“The lack of annotated publicly-available medical images is a major barrier for innovations,” the researchers wrote in Nature Medicine. “At the same time, many de-identified images and much knowledge are shared by clinicians on public forums such as medical Twitter.”
In this case, the goal “is to train a model that can understand both the visual image and the text description,” Zou said in the SCOPE blog post.
“Pathology is perhaps even more suited to Twitter than many other medical fields because for most pathologists, the bulk of our daily work revolves around the interpretation of images for the diagnosis of human disease,” wrote Jerad M. Gardner, MD, a dermatopathologist and section head of bone/soft tissue pathology at Geisinger Medical Center in Danville, Pa., in a blog post about the Pathology Hashtag Ontology project. “Twitter allows us to easily share images of amazing cases with one another, and we can also discuss new controversies, share links to the most cutting edge literature, and interact with and promote the cause of our pathology professional organizations.”
The researchers used the 32 subspecialty hashtags to retrieve English-language tweets posted from 2006 to 2022. Images in the tweets were “typically high-resolution views of cells or tissues stained with dye,” according to the SCOPE blog post.
The researchers collected a total of 232,067 tweets and 243,375 image-text pairs across the 32 subspecialties, they reported. They augmented this with 88,250 replies that received the highest number of likes and had at least one keyword from the ICD-11 codebook. The SCOPE blog post noted that the rankings by “likes” enabled the researchers to screen for high-quality replies.
They then refined the dataset by removing duplicates, retweets, non-pathology images, and tweets marked by Twitter as being “sensitive.” They also removed tweets containing question marks, as this was an indicator that the practitioner was asking a question about an image rather than providing a description, the researchers wrote in Nature Medicine.
They cleaned the text by removing hashtags, Twitter handles, HTML tags, emojis, and links to websites, the researchers noted.
The final OpenPath dataset included:
116,504 image-text pairs from Twitter posts,
59,869 from replies, and
32,041 image-text pairs scraped from the internet or obtained from the LAION dataset.
The latter is an open-source database from Germany that can be used to train text-to-image AI software such as Stable Diffusion.
Training the PLIP AI Platform
Once they had the dataset, the next step was to train the PLIP AI model. This required a technique known as contrastive learning, the researchers wrote, in which the AI learns to associate features from the images with portions of the text.
As explained in Baeldung, an online technology publication, contrastive learning is based on the idea that “it is easier for someone with no prior knowledge, like a kid, to learn new things by contrasting between similar and dissimilar things instead of learning to recognize them one by one.”
“The power of such a model is that we don’t tell it specifically what features to look for. It’s learning the relevant features by itself,” Zou said in the SCOPE blog post.
The resulting AI PLIP tool will enable “a clinician to input a new image or text description to search for similar annotated images in the database—a sort of Google Image search customized for pathologists,” SCOPE explained.
“Maybe a pathologist is looking at something that’s a bit unusual or ambiguous,” Zou told SCOPE. “They could use PLIP to retrieve similar images, then reference those cases to help them make their diagnoses.”
The Stanford University researchers continue to collect pathology images from X. “The more data you have, the more it will improve,” Zou said.
Pathologists will want to keep an eye on the Stanford Medicine research team’s progress. The PLIP AI tool may be a boon to diagnostics and improve patient outcomes and care.
Already-existing clinical laboratory blood test may be new standard for detecting Alzheimer’s biomarkers
In Sweden, an independent study of an existing blood test for Alzheimer’s disease—called ALZpath—determined that this diagnostic assay appears to be “just as good as, if not surpass, lumbar punctures and expensive brain scans at detecting signs of Alzheimer’s in the brain,” according to a report published by The Guardian.
Alzheimer’s disease is one of the worst forms of dementia and it affects more than six million people annually according to the Alzheimer’s Association. Clinical laboratory testing to diagnose the illness traditionally involves painful, invasive spinal taps and brain scans. For that reason, researchers from the University of Gothenburg in Sweden wanted to evaluate the performance of the ALZpath test when compared to these other diagnostic procedures.
Motivated to seek a less costly, less painful, Alzheimer’s biomarker for clinical laboratory testing, neuroscientist Nicholas Ashton, PhD, Assistant Professor of Neurochemistry at the University of Gothenburg, led a team of scientists that looked at other common biomarkers used to identify changes in the brain of Alzheimer’s patients. That led them to tau protein-based blood tests and specifically to the ALZpath blood test for Alzheimer’s disease developed by ALZpath, Inc., of Carlsbad, Calif.
In their JAMA article, they wrote, “the pTau217 immunoassay showed similar accuracies to cerebrospinal fluid biomarkers in identifying abnormal amyloid β (Aβ) and tau pathologies.”
In an earlier article published in medRxiv, Ashton et al wrote, “Phosphorylated tau (pTau) is a specific blood biomarker for Alzheimer’s disease (AD) pathology, with pTau217 considered to have the most utility. However, availability of pTau217 tests for research and clinical use has been limited.”
Thus, the discovery of an existing pTau217 assay (ALZpath) that is accessible and affordable is a boon to Alzheimer’s patients and to the doctors who treat them.
“The ALZpath pTau217 assay showed high diagnostic accuracy in identifying elevated amyloid (AUC, 0.92-0.96; 95%CI 0.89-0.99) and tau (AUC, 0.93-0.97; 95%CI 0.84-0.99) in the brain across all cohorts. These accuracies were significantly higher than other plasma biomarker combinations and equivalent to CSF [cerebrospinal fluid] biomarkers,” an ALZpath press release noted.
“This is an instrumental finding in blood-based biomarkers for Alzheimer’s, paving the way for the clinical use of the ALZpath pTau217 assay,” stated Henrik Zetterberg, MD, PhD (above), Professor of Neurochemistry at the University of Gothenburg and co-author of the study. “This robust assay is already used in multiple labs around the globe.” Clinical laboratories may soon be receiving doctors’ orders for pTau217 blood tests for Alzheimer’s patients. (Photo copyright: University of Gothenburg.)
Study Details
Ashton’s team conducted a cohort study that “examined data from three single-center observational cohorts.” The cohorts included:
“Participants included individuals with and without cognitive impairment grouped by amyloid and tau (AT) status using PET or CSF biomarkers. Data were analyzed from February to June 2023,” the researchers wrote.
These trials from the US, Canada, and Spain featured 786 participants and featured “either a lumbar puncture or an amyloid PET scan to identify signs of amyloid and tau proteins—hallmarks of Alzheimer’s disease,” The Guardian reported, adding that results of the University of Gothenburg’s study showed that the ALZpath pTau217 blood test “was superior to brain atrophy assessments, in identifying signs of Alzheimer’s.”
“80% of individuals could be definitively diagnosed on a blood test without any other investigation,” Ashton told The Guardian.
Diagnosis Needed to Receive Alzheimer’s Disease Treatments
“If you’re going to receive [the new drugs], you need to prove that you have amyloid in the brain,” Ashton told The Guardian. “It’s just impossible to do spinal taps and brain scans on everyone that would need it worldwide. So, this is where the blood test [has] a huge potential.”
Even countries where such drugs were not yet available (like the UK) would benefit, Ashton said, because the test, “Could potentially say that this is not Alzheimer’s disease and it could be another type of dementia, which would help to direct the patient’s management and treatment routine.”
However, Ashton himself noted the limitations of the new findings—specifically that there is no success shown yet in Alzheimer’s drugs being taken by symptom-free individuals.
“If you do have amyloid in the brain at 50 years of age, the blood test will be positive,” he said. “But what we recommend, and what the guidelines recommend with these blood tests, is that these are to help clinicians—so someone must have had some objective concern that they have Alzheimer’s disease, or [that] their memory is declining,” he told The Guardian.
Experts on the Study Findings
“Blood tests could be used to screen everyone over 50-years old every few years, in much the same way as they are now screened for high cholesterol,” David Curtis, MD, PhD, Honorary Professor in the Genetics, Evolution and Environment department at University College London, told The Guardian.
“Results from these tests could be clear enough to not require further follow-up investigations for some people living with Alzheimer’s disease, which could speed up the diagnosis pathway significantly in future,” Richard Oakley, PhD, Associate Director of Research and Innovation at the Alzheimer’s Society, UK, told The Guardian.
Though Oakley found the findings promising, he pointed out what should come next. “We still need to see more research across different communities to understand how effective these blood tests are across everyone who lives with Alzheimer’s disease,” he said.
“Expanding access to this highly accurate Alzheimer’s disease biomarker is crucial for wider evaluation and implementation of AD blood tests,” the researchers wrote in JAMA Neurology.
“ALZpath makers are in discussions with labs in the UK to launch it for clinical use this year, and one of the co-authors, Henrik Zetterberg, MD, PhD, Professor of Neurochemistry at the University of Gothenburg, is making the assay available for research use as part of the ‘biomarker factory’ at UCL,” The Guardian reported.
In the US, to be prescribed any of the available Alzheimer’s medications, a doctor must diagnose that the patient has amyloid in the brain. A pTau217 diagnostic blood test could be used to make such a diagnosis. Currently, however, the test is only available “for research studies through select partner labs,” Time reported.
“But later this month, doctors in the US will be able to order the test for use with patients. (Some laboratory-developed tests performed by certain certified labs don’t require clearance from the US Food and Drug Administration.),” Time added.
It may be that the University of Gothenburg study will encourage Alzheimer’s doctors in the UK and around the world to consider ordering pTau217 diagnostic blood tests from clinical laboratories, rather than prescribing spinal taps and brains scans for their Alzheimer’s patients.
HHS Office of Inspector General was the latest to examine the quality control problems that led to distribution of inaccurate test to clinical laboratories nationwide
Failure on the part of the Centers for Disease Control and Prevention (CDC) to produce accurate, dependable SARS-CoV-2 clinical laboratory test kits at the start of the COVID-19 pandemic continues to draw scrutiny and criticism of the actions taken by the federal agency.
In the early weeks of the COVID-19 pandemic, the CDC distributed faulty SARS-CoV-2 test kits to public health laboratories (PHLs), delaying the response to the outbreak at a critical juncture. That failure was widely publicized at the time. But within the past year, two reports have provided a more detailed look at the shortcomings that led to the snafu.
“We identified weaknesses in CDC’s COVID-19 test kit development processes and the agencywide laboratory quality processes that may have contributed to the failure of the initial COVID-19 test kits,” the OIG stated in its report.
Prior to the outbreak, the agency had internal documents that were supposed to provide guidance for how to respond to public health emergencies. However, “these documents do not address the development of a test kit,” the OIG stated.
“If the CDC can’t change, [its] importance in health in the nation will decline,” said microbiologist Jill Taylor, PhD (above), Senior Adviser for the Association of Public Health Laboratories in Washington, DC. “The coordination of public health emergency responses in the nation will be worse off.” Clinical laboratories that were blocked from developing their own SARS-CoV-2 test during the pandemic would certainly agree. (Photo copyright: Columbia University.)
Problems at the CDC’s RVD Lab
Much of the OIG’s report focused on the CDC’s Respiratory Virus Diagnostic (RVD) lab which was part of the CDC’s National Center for Immunization and Respiratory Diseases (NCIRD). The RVD lab had primary responsibility for developing, producing, and distributing the test kits. Because it was focused on research, it “was not set up to develop and manufacture test kits and therefore had no policies and procedures for developing and manufacturing test kits,” the report stated.
The RVD lab also lacked the staff and funding to handle test kit development in a public health emergency, the report stated. As a result, “the lead scientist not only managed but also participated in all test kit development processes,” the report stated. “In addition, when the initial test kit failed at some PHLs, the lead scientist was also responsible for troubleshooting and correcting the problem.”
To verify the test kit, the RVD lab needed samples of viral material from the agency’s Biotechnology Core Facility Branch (BCFB) CORE Lab, which also manufactured reagents for the kit.
“RVD Lab, which was under pressure to quickly create a test kit for the emerging health threat, insisted that CORE Lab deviate from its usual practices of segregating these two activities and fulfill orders for both reagents and viral material,” the report stated.
This increased the risk of contamination, the report said. An analysis by CDC scientists “did not determine whether a process error or contamination was at fault for the test kit failure; however, based on our interviews with CDC personnel, contamination could not be ruled out,” the report stated.
The report also cited the CDC’s lack of standardized systems for quality control and management of laboratory documents. Labs involved in test kit development used two different incompatible systems for tracking and managing documents, “resulting in staff being unable to distinguish between draft, obsolete, and current versions of laboratory procedures and forms.”
Outside Experts Weigh In
The OIG report followed an earlier review by the CDC’s Laboratory Workgroup (LW), which consists of 12 outside experts, including academics, clinical laboratory directors, state public health laboratory directors, and a science advisor from the Association of Public Health Laboratories. Members were appointed by the CDC Advisory Committee to the Director.
This group cited four major issues:
Lack of adequate planning: For the “rapid development, validation, manufacture, and distribution of a test for a novel pathogen.”
Ineffective governance: Three labs—the RVD Lab, CORE Lab, and Reagent and Diagnostic Services Branch—were involved in test kit development and manufacturing. “At no point, however, were these three laboratories brought together under unified leadership to develop the SARS-CoV-2 test,” the report stated.
Poor quality control and oversight: “Essentially, at the start of the pandemic, infectious disease clinical laboratories at CDC were not held to the same quality and regulatory standards that equivalent high-complexity public health, clinical and commercial reference laboratories in the United States are held,” the report stated.
Poor test design processes: The report noted that the test kit had three probes designed to bind to different parts of the SARS-CoV-2 nucleocapsid gene. The first two—N1 (topology) and N2 (intracellular localization)—were designed to match SARS-CoV-2 specifically, whereas the third—N3 (functions of the protein)—was designed to match all Sarbecoviruses, the family that includes SARS-CoV-2 as well as the coronavirus responsible for the 2002-2004 SARS outbreak.
The N1 probe was found to be contaminated, the group’s report stated, while the N3 probe was poorly designed. The report questioned the decision to include the N3 probe, which was not included in European tests.
Also lacking were “clearly defined pass/fail threshold criteria for test validation,” the report stated.
Advice to the CDC
Both reports made recommendations for changes at the CDC, but the LW’s were more far-reaching. For example, it advised the agency to establish a senior leader position “with major responsibility and authority for laboratories at the agency.” This individual would oversee a new Center that would “focus on clinical laboratory quality, laboratory safety, workforce training, readiness and response, and manufacturing.”
In addition, the CDC should consolidate its clinical diagnostic laboratories, the report advised, and “laboratories that follow a clinical quality management system should have separate technical staff and space from those that do not follow such a system, such as certain research laboratories.”
The report also called for collaboration with “high functioning public health laboratories, hospital and academic laboratories, and commercial reference laboratories.” For example, collaborating on test design and development “should eliminate the risk of a single point of failure for test design and validation,” the LW suggested.
CBS News reported in August that the CDC had already begun implementing some of the group’s suggestions, including agencywide quality standards and better coordination with state labs.
However, “recommendations for the agency to physically separate its clinical laboratories from its research laboratories, or to train researchers to uphold new quality standards, will be heavy lifts because they require continuous funding,” CBS News reported, citing an interview with Jim Pirkle, MD, PhD, Director, Division of Laboratory Sciences, National Center for Environmental Health, at the CDC.
By emphasizing HPV vaccinations while having clinical laboratories continue to perform Pap smears, Australia’s rate of cervical cancer has dropped notably
There is currently a global push to completely eradicate cervical cancer and Australia is leading the way with increased funding. It is also focusing on hard-to-reach and underserved populations. Australia is hoping to be first in the world to accomplish this feat by 2035.
For a number of decades, the Pap smear has been the primary screening tool for cervical cancer, as most pathologists and clinical laboratory managers know. However, today it plays a lesser role due to the effectiveness of HPV (human papillomavirus) diagnostic testing, which was put into cervical cancer screening guidelines in 2004.
Then came the first HPV vaccine in 2006. Australia was one of the first nations to implement HPV vaccination programs. By 2010, Australia was working to vaccinate every child. Now, 14 years later, the pool of adults vaccinated against HPV in that nation is causing the rates of cervical cancer to fall.
That means much less cervical cancer test volume for cytotechnologists and cytopathologists, freeing them up to devote their skills to other diagnostic tests.
As the country continues to funnel resources into hitting a zero cancer status, the additional drive will “connect Australia’s world-leading cervical cancer expertise with governments across the region to get HPV vaccine programs up and running, expand screening and treatment, and build health workforce capacity,” said Australia’s Minister for Foreign Affairs office in a press release.
“Australia has always punched above its weight when it comes to cervical cancer, and now Australia is on track to be the first country in the world to eliminate this deadly disease,” said Hon Ged Kearney, MP, RN (above), Assistant Minister for Health and Aged Care and a member of the government’s House of Representatives, in a press release. “By supporting the Pacific and Southeast Asia region [to] eliminate cervical cancer, we are another step closer to ridding the world of this disease.” Clinical laboratories and cytopathologists may soon see less reliance on Pap smears for screening and a shift toward HPV vaccinations to lower the rate of cervical cancer in the US as well. (Photo copyright: Australian Labor Party.)
90% of eligible people will be vaccinated against HPV (including girls and boys).
70% of eligible people will be screened every five years.
95% of eligible people will receive the best possible treatment for precancer and cancer.
In addition to $48.2 million in funding over four years, the program provides:
On the spot testing of samples in First Nations [aka, First Peoples] communities, allowing immediate follow up.
Support for nurses, First Nations health practitioners, and midwives to request pathology for cervical screening.
Increasing support for GPs to undertake colposcopies.
Helping the Underserved
Reaching a wider audience is a large part of Australia’s focus.
“One of my priorities is to address inequities in our health system. I want to make sure that everyone can get access to screening—and all healthcare—no matter where [they] live,” Kearney added. Among the populations sought are First Nations, LGBTIQA+, disabled individuals, and those living away from large cities.
“$8.3 million has been allocated to implement innovate screening models to support such communities,” the Minister for Foreign Affairs office noted in the press release.
Meeting people where they are, and reaching underserved populations, can make a huge difference, especially considering how cervical cancer affects these people. “First Nations women are almost twice as likely to be diagnosed with cervical cancer and face significant barriers to participating in cervical screening compared to non-indigenous women,” the press release notes.
“These tests allow privacy and help to break down barriers for thousands of people who have never screened—including women who have experienced sexual violence, LGBTIQA+ people, and culturally and linguistically diverse and First Nations communities,” the Minister for Foreign Affairs office stated.
There is hope that the push will cause a great shift to other underserved communities as well.
“A quarter of global cervical cancer cases occur in our region, the Indo-Pacific. Tragically, in the Pacific, women are dying at up to 13 times the rate of women in Australia,” said Penny Wong, Australian Minister for Foreign Affairs, in the press release.
How the US Fares in Cervical Cancer Vaccinations
Australia’s vaccination rates far exceed those in the United States. The US government currently recommends HPV vaccination between the ages of 11-12 years old, though it could be administered starting at age nine.
“HPV vaccination is recommended for all persons through age 26 years who were not adequately vaccinated earlier,” the NIH’s National Cancer Institute (NCI) reports.
For years the standard focus for cervical cancer screening has been on the Pap smear. Data show the US lags behind many countries on the rate of HPV vaccination. NCI data show that, as of 2021, in the US just 58.5% of 13-15 year-olds “had received two or three doses of HPV vaccine as recommended,” NCI reported.
With the US’s standard of care still focused on the Pap smear, patients are beginning their cervical cancer prevention journey at a later age. This is because the preliminary age to get a Pap smear in the US is 21 years old, with follow-up exams every three years, the NCI reported.
Even those in this country who are sexually active are not recommended to get screening earlier than 21.
The NCI recommends HPV testing every five years starting at age 30 until 65, with Pap tests every three years.
Clinical laboratories may soon find that, while the US has been slower to get on board with HPV vaccinations, trends in other nations indicate that this may soon change. The reliance that was once placed on the Pap smears prior to 2000 will likely give way to HPV vaccinations at ages and vaccination rates that mirror programs in countries like Australia—where marked reductions in the rate of cervical cancer demonstrate the effectiveness of a successful HPV vaccination program.
Immunotherapy device could also enable clinical laboratories to receive in vivo biomarker data wirelessly
Researchers from Rice University in Houston and seven other states in the US are working on a new oncotherapy sense-and-respond implant that could dramatically improve cancer outcomes. Called Targeted Hybrid Oncotherapeutic Regulation (THOR), the technology is intended primarily for the delivery of therapeutic drugs by monitoring specific cancer biomarkers in vivo.
Through a $45 million federal grant from the Advanced Research Projects Agency for Health (ARPA-H), the researchers set out to develop an immunotherapy implantable device that monitors a patient’s cancer and adjusts antibody treatment dosages in real time in response to the biomarkers it measures.
It’s not a far stretch to envision future versions of the THOR platform also being used diagnostically to measure biomarker data and transmit it wirelessly to clinical laboratories and anatomic pathologists.
ARPH-A is a federal funding agency that was established in 2022 to support the development of high-impact research to drive biomedical and health breakthroughs. THOR is the second program to receive funding under its inaugural Open Broad Agency Announcement solicitation for research proposals.
“By integrating a self-regulated circuit, the THOR technology can adjust the dose of immunotherapy reagents based on a patient’s responses,” said Weiyi Peng, MD, PhD (above), Assistant Professor of Biology and Biochemistry at the University of Houston and co-principal investigator on the research, in a UH press release. “With this new feature, THOR is expected to achieve better efficacy and minimize immune-related toxicity. We hope this personalized immunotherapy will revolutionize treatments for patients with peritoneal cancers that affect the liver, lungs, and other organs.” If anatomic pathologists and clinical laboratories could receive biometric data from the THOR device, that would be a boon to cancer diagnostics. (Photo copyright: University of Houston.)
Antibody Therapy on Demand
Omid Veiseh, PhD, Associate Professor of Bioengineering at Rice University and principal investigator on the project, described the THOR device as a “living drug factory” inside the body. The device is a rod-like gadget that contains onboard electronics and a wireless rechargeable battery. It is three inches long and has a miniaturized bioreactor that contains human epithelial cells that have been engineered to produce immune modulating therapies.
“Instead of tethering patients to hospital beds, IV bags, and external monitors, we’ll use a minimally invasive procedure to implant a small device that continuously monitors their cancer and adjusts their immunotherapy dose in real time,” said Veiseh in a Rice University press release. “This kind of ‘closed-loop therapy’ has been used for managing diabetes, where you have a glucose monitor that continuously talks to an insulin pump.
But for cancer immunotherapy, it’s revolutionary.”
The team believes the THOR device will have the ability to monitor biomarkers and produce an antibody on demand that will trigger the immune system to fight cancer locally. They hope the sensor within THOR will be able to monitor biomarkers of toxicity for the purpose of fine-tuning therapies to a patient immediately in response to signals from a tumor.
“Today, cancer is treated a bit like a static disease, which it’s not,” Veiseh said. “Clinicians administer a therapy and then wait four to six weeks to do radiological measurements to see if the therapy is working. You lose quite a lot of time if it’s not the right therapy. The tumor may have evolved into a more aggressive form.”
The THOR device lasts 60 days and can be removed after that time. It is designed to educate the immune system to recognize a cancer and prevent it from recurring. If the cancer is not fully eradicated after the first implantation, the patient can be implanted with THOR again.
Use of AI in THOR Therapy
The researchers plan to spend the next two and a half years building prototypes of the THOR device, testing them in rodents, and refining the list of biomarkers to be utilized in the device. Then, they intend to take an additional year to establish protocols for the US Food and Drug Administration’s (FDA) good manufacturing practices requirements, and to test the final prototype on large animals. The researchers estimate the first human clinical trials for the device will begin in about four years.
“The first clinical trial will focus on refractory recurrent ovarian cancer, and the benefit of that is that we have an ongoing trial for ovarian cancer with our encapsulated cytokine ‘drug factory’ technology,” said Veiseh in the UH press release.
The group is starting with ovarian cancer because research in this area is lacking and it will provide the opportunity for THOR to activate the immune system against ovarian cancer, which is typically challenging to fight with immunotherapy approaches. If successful in ovarian cancer, the researchers hope to test THOR in other cancers that metastasize within the abdomen, such as:
All control and decision-making will initially be performed by a healthcare provider based on signals transmitted by THOR using a computer or smartphone. However, Veiseh sees the device ultimately being powered by artificial intelligence (AI) algorithms that could independently make therapeutic decisions.
“As we treat more and more patients [with THOR], the devices are going to learn what type of biomarker readout better predicts efficacy and toxicity and make adjustments based on that,” he predicted. “Between the information you have from the first patient versus the millionth patient you treat, the algorithm is just going to get better and better.”
Moving Forward
In addition to UH and Rice University, scientists working on the project come from several institutions, including:
More research and clinical trials are needed before THOR can be used in the clinical treatment of cancer patients. If the device reaches the commercialization stage, Veiseh plans to either form a new company or license the technology to an existing company for further development.
“We know that the further we advance it in terms of getting that human data, the more likely it is that this could then be transferred to another entity,” he told Precision Medicine Online.
Pathologists and clinical laboratories will want to monitor the progress of the THOR technology’s ability to sense changes in cancer biomarkers and deliver controlled dosages of antibiotic treatments.
This is the second of a three-part series on revenue cycle management for molecular testing laboratories and pathology practices, produced in collaboration with XiFin, Inc.
Second of a 3-part series, this article will detail what molecular diagnostics and pathology groups need to understand about coding, billing, and denial management to maximize revenue and cash flow successfully.
In the first article, we discussed how molecular diagnostics and pathology groups can enhance the patient experience, physician engagement, and payer relations. Now, we will detail how denial management can successfully maximize revenue and cash flow. As we discussed in the last article, revenue cycle management (RCM) is much more than billing.
Today’s rapidly changing environment of directives and expectations from payers, patients, and health systems require deeper understanding, great agility, and strategy in every aspect of business. Creating opportunities to provide better service, adopt state-of-the-art technologies, and build robust processes and partnerships can make or break the long-term success of a laboratory or pathology practice.
Technical assessments are often required to establish clinical validity and utility to achieve payer coverage for novel genetic tests. Achieving payer coverage requires a deep understanding of how-to code tests compliantly and to facilitate reimbursement.
“We recommend that molecular diagnostics laboratories consult with coding experts to fully understand the coding requirements for each genetic test,” says Clarisa Blattner, XiFin Senior Director, Revenue and Payor Optimization. “Ensuring reimbursement requires knowing payer policies and to track denial trends by payer over time to identify changes.”
Blattner noted that payer policies and behavior are constantly changing. Labs, and their billing partners must stay abreast of changes to avoid lengthy delays that denials and subsequent appeals can cause. Understanding the documentation that is required with claims is invaluable. Knowing these requirements up front and submitting complete claims with all required medical records and documentation of medical necessity goes a long way toward facilitating reimbursement.
Payers are adopting increasingly rigid policies that are often inconsistent with others. Reimbursements continue to be cut while quality reporting requirements rise.
Diagnostics laboratories that conduct genetic testing must also overcome four common challenges:
Achieving and expanding payer coverage with coverage determination that defines reasonable and medically necessary services and tests.
Knowing how to code the tests correctly with medical nomenclature to report services and/or tests to a payer.
Ensuring payment/reimbursement for services/tests based on services/tests rendered and coverage determination.
Maintaining compliance and keeping abreast of billing compliance and having a voice in reform
“We also recommend that laboratories conduct internal audits that reconcile laboratory information system (LIS) data with RCM system data,” Blattner continued. “Labs with a robust business intelligence (BI) solution can proactively identify outliers, such as accessions that exist in one system but not the other.”
Maintain Your Billing System and Maximize Clean Claim Submissions
Laboratories should be sure that these four payer services are being handled appropriately, whether it is by the lab or an RCM partner:
1. Payer relations: An effective payer relations team monitors denials and coordinates with payers. This team reviews front-end payer rejections, coordinates with clients (i.e., ordering physicians), and identifies and updates edits based on payer policies and behavior changes.
2. Electronic data interchange (EDI) enrollment: This team handles monitoring and proactive enrollment for electronic submissions and helps ensure bidirectional transaction automation.
3. EDI analysts: Experts in healthcare EDI who investigate errors, participate in standards development and testing, as well as payer education and coordination.
4. EDI operations: These specialized technicians configure files and ensure the reconciliation of claim-level submissions.
Efficiently Upload and Store Medical Records and Documentation
Although laboratories do not directly control patient medical records, it is essential to understand that diagnosis codes alone are generally insufficient.
Laboratory sales representatives must work with clients and ordering physicians to ensure medical records have all the information required for payment. Ensuring that the payers expedite payment requires efficient uploading and storing of medical records and documentation:
Align with payers on clinical utility evidence requirements, current billing policies, and preferred coding approach.
Leverage the support and advocacy of key opinion leaders (KOLs).
Collaborate with clinicians on the prior authorization process.
Select an RCM partner that helps you maximize process automation and front-end edits.
Leverage a business intelligence (BI) system that simplifies the tracking of key performance indicators (KPIs), helps identify payer policy and behavior changes early, and highlights changes in key business trends.
The RCM system must be able to upload and store medical records and documentation. The required medical information typically includes the following:
Who? Ordering/referring provider.
What? What service(s)/test(s) is/are being ordered?
Where? Where is the specimen being sent?
When? What is the date of service (DOS)?
Why? What are the patient’s signs/symptoms, or what prompted the test to be ordered?
How? How are the test results used to manage the patient’s medical condition?
But even after including all of the correct medical information, denials are inevitable. There are important steps labs can take to streamline denial management.
The Importance of Patient Engagement in Maximizing Reimbursement
Patient engagement plays an essential role in facilitating reimbursement and maximizing cash collection. Patients expect transparency and ease of information access from their healthcare encounters, just as they experience in all other areas of their lives. Because most laboratory, pathology, and molecular encounters are not directly patient-facing, proven payment accelerating engagement tools are essential. Dynamic portals, electronic statements, and text messages are essential, especially when it comes to communication regarding errors and patient financial responsibility.
XiFin customer data show a substantive increase in patient payments received in the first 30 days of the dunning cycle after integrating texting and automated calls into the traditional process. For example, a XiFin customer collected 26.6% more of the revenue in the first 30 days after implementing a text reminder between the first and second paper statements. Prior to implementation, the customer followed a traditional three-statement dunning cycle:
42.6% of total payments received occurred after the first statement (within the first 29 days of the dunning cycle).
34.8% occurred after sending the second statement (between days 30-59 of the dunning cycle).
22.6% were received after sending the third and final statement (during days 60-90 of thedunning cycle).
The convenience of text messaging allows patients to connect to the call center or to the patient portal, where a payment can be made immediately. XiFin customers can customize their dunning cycle, depending on how their specific patient population responds to texts, paper statements, and the timing between billing cycles. Studying the behaviors of patient interactions at the client level, rather than only referencing the status quo of macro-level trending, empowers a more strategic approach to engagement and improving overall patient satisfaction.
Denial Trends Driving Reduced Revenue and Higher Costs
Denials extend time in accounts receivable, contributing to bad debt on services already rendered and laboratory expenses absorbed. Denials also often require the most attention from staff – increasing the cost of billing. Hard denials, such as Medical Necessity, make up the most challenging revenue to collect, comprising about 5-10% of total denials received. In addition to creating delays and revenue loss, denials illustrate how payers administer their policies, even when those policies are unpublished.
Fundamentally, an effective RCM process is rooted in the ability to file clean claims to the degree that is possible. Improving those outcomes requires focus on the exceptions – the dirty claims – the denials.
“At XiFin, we invest in front-end configurations and workflows to catch denials prior to submitting the claim to the payer,” continued Blattner. “As we monitor denial trends, we build more robust front-end workflows and add automation (such as integrating with insurance discovery and prior authorization vendors) to reduce the associated burden on billing teams.”
In addition, molecular testing coverage continues to expand, reducing non-covered denials. The stabilization of these medical policy-related denials is positive. The jump in demographic denials, however, requires additional consideration.
Paid vs. Denied by Payer Group
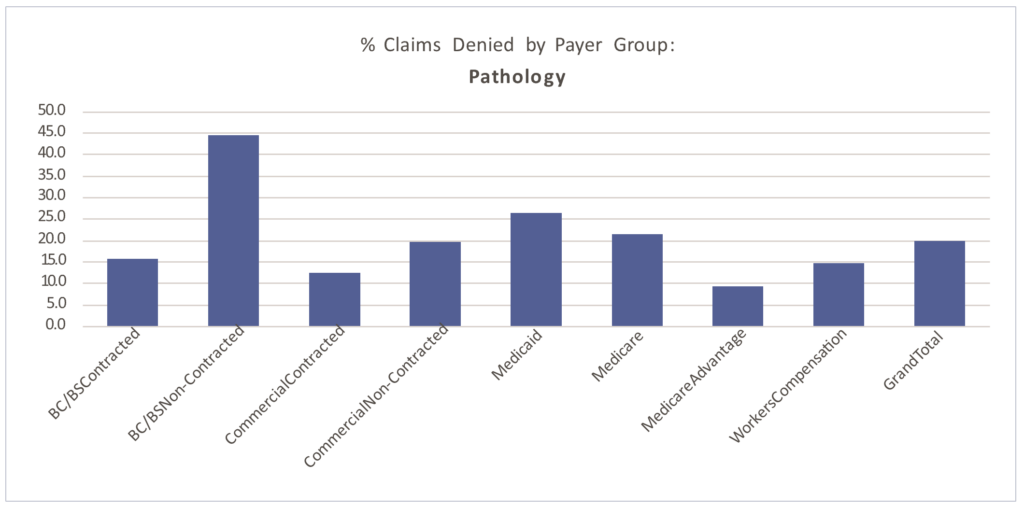
Denial patterns vary among payers. The percentage of claims denied also differs by segment, largely due to the type of testing performed.
Of the claims XiFin processes annually (approximately $50 billion in charges), 22.5% are denied. The graphs below demonstrate molecular testing’s higher propensity for denial (27.5% of claims billed), driven by non-covered, medical necessity, and prior authorization requirement challenges.
Routine pathology has closer to a 20% denial rate overall. The average percentage of billed claims that are denied by segment are:
■ Clinical: 13.62%
■ Molecular: 27.19%
■ Pathology: 19.82%
Molecular testing has a higher propensity for denial (27.5% of claims billed), driven by non-covered, medical necessity, and prior authorization requirement challenges. Routine pathology has closer to a 20% denial rate overall.
Clinical laboratory denial rates averaged 13.62% in 2021. As seen in the table below,clinical laboratories saw a significant decline in experimental/investigational denials between 2018 and 2021.
Denial Type
Molecular % of Total Denied 2018
Clinical % of Total Denied 2021
Variance (% change 2021 vs. 2018)
Benefit Maximum Reached
39.3%
29.7%
-24.4%
Claim Specific Negotiated Discount
17.6%
18.1%
2.8%
Coordination of Benefits
4.1%
16.3%
298%
Coverage Terminated
6.6%
13.4%
103%
Diagnosis Not Covered
11.3%
6.4%
-43.4%
Duplicate Denial
8.3%
3.4%
-57.8%
Experimental Investigational
0.1%
2.7%
2600%
HSA
2.1%
2.4%
14.3%
Incorrect Payer
0.9%
1.6%
77.8%
Non-Covered
2.2%
1.1%
-50.0%
Patient Cannot be Identified
0.7%
0.8%
14.3%
Patient Enrolled in Hospice
0.5%
0.5%
0.0%
Prior Authorization
0.2%
0.2%
0.0%
Procedure Code Inconsistent with the Modifier Used or a Required Modifier is Missing
1.6%
0.1%
-87.5%
Procedure Not Paid Separately
0.5%
0.1%
-60.0%
Service Not Payable per Managed Care Contract
0.1%
0.0%
-100%
Molecular claims continue to experience the highest denial rates of any laboratory segment. With an average denial rate of 27%, molecular continues to be arevenue recovery workflow heavy on the back-end. As a percentage of the total denial population, between 2018 and 2021, XiFin experienced increases in patient-coverage denials, such as coordination of benefits (298%), coverage terminated (103%), and experimental/investigational (2600%). Decreases in diagnosis not covered denials (-43.4%) and duplicate denials (-57.8%) are also recognized.
Exome/Genome Testing must be administered by specialized technicians with specificcredentials, creating potential backlogs. They can take 8, 12, or even 16 weeks to complete, depending on testing methodologies. This presents a high risk of timely filing denials for the many payers that have adopted 90-day timely filing limits. XiFin recommended practice: Explore amending your payer contracts to extend timely filing limits on these tests.
Denial Type
Pathology % of Total Denied 2018
Pathology % of Total Denied 2021
Variance (% change 2021 vs. 2018)
Prior Authorization
28.9%
36.1%
24.6%
Duplicate Denial
21.5%
21.2%
-1.9%
Non-Covered
14.1%
10.1%
-27.7%
Services Not Prov. By Network/Primary Care Provider
8.8%
8.5%
-3.4%
Procedure Not Paid Separately
4.4%
5.1%
15.9%
Services Not Authorized by Network/Primary Care Provider
3.6%
3.8%
5.6%
Procedure Code Inconsistent with the Modifier Used or a Required Modifier is Missing
1.5%
3.3%
120%
Coverage Terminated
2.2%
2.6%
18.2%
Coordination of Benefits
3.8%
2.4%
-34.2%
Patient Cannot Be Identified
3.1%
2.3%
-25.8%
Remark Code
5.9%
2.1%
-64.4%
Experimental Investigational
1.0%
1.2%
20.0%
Benefit Maximum Reached
0.4%
1.0%
175%
Patient Enrolled in Hospice
0.4%
0.1%
-75.0%
Incorrect Payer
0.0%
0.1%
100%
Service Not Payable per Managed Care Contract
0.2%
0.0%
-100%
Anatomic pathology denials have increased by approximately 5% from 2018 to 2021.As a percentage of the total denial population, the lack of prior authorization is the highestcontributor to this increase, having grown 24.6%. There was an increase inprocedure code inconsistent with modifier denials (120% increase) and a decreasein non-covered denials (-27.7%).
Importance of an Efficient and Effective Appeals Process
Front-end edits and configurations help mitigate backend denials. Capturing potential denial-related issues proactively are the most effective way to maintain a manageable AR and improve the propensity to pay. For example, payers that observe National Correct Coding Initiative (NCCI) and Medically Unlikely Edits (MUEs) will consider all Current Procedural Terminology (CPT) codes billed for that patient for the same Date of Service (DOS), even when not billed on the same claim form.
Denials are inevitable if your current billing process does not have edits in place to perform a historical review of charges for the same patient on the same DOS.
Denials are unavoidable, and not all known issues can be addressed on the front end of the process. An example of this is denial code CO252, which is an additional information denial. It indicates the payer is requesting additional documentation (i.e., clinical information, medical records, and test results) before issuing payment – essentially performing an audit to ensure the services billed are reasonable and necessary and medical necessity is justified and documented.
“These are not always complex molecular tests; they can be routine pathology claims,” said Blattner “Each time we receive a CO252 denial it has to be appealed with additional documentation found in the patient’s medical records. Though it is inevitable, we must wait on the denial before we can take action.”
Segment
Appeal-Payments as % of Total Insurance Payments Received
Average Payment Amount per Appeal
Clinical
0.11%
$121
Molecular
6.56%
$1,420
Pathology
1.12%
$327
Industry Average
3.39%
$623
Payment collection per appeal continues to be stable in the pathology (averaging 1-2%) and clinical segments, where appeals are less prolific. Revenue recovered by corrected claims is excluded since these claims follow a separate process and impact denial codes such as CO97 (Procedure or service isn’t paid for separately), CO18 (Duplicate), and CO234 (Procedure not paid separately). Further, a single appeal process is not sufficient. A robust appeals process here becomes critical. Specifically in molecular testing, appeals carry a heightening impact on revenue collection. In 2020, appeals accounted for 5% of the total revenue generated by XiFin customers. In 2021, that increased to 6.5%.
Appeal Success Rates by Payer Group by Segment
The next four charts show appeal success rates by payer group for 2021, overall and by market segment for clinical, molecular, and pathology. The fifth chart illustrates the incremental impact of multiple appeal attempts by market segment.
This assessment only includes activity related to revenue recovery through an appeals process. Some denials can be addressed by filing of a corrected claim and can be a much more efficient process. Although ideal, corrected claims are not always possible, depending on denial type and individual payer preferences.
% of Total Appeals Filed
% of AppealsPaid after 1st Attempt
% of AppealsPaid after 2nd Attempt
% of AppealsPaid after 3rd Attempt
Avg Paymentper Appeal
Clinical
17.4%
17.8%
9.9%
$ 276
Additional Information
70.1%
20.9%
20.3%
10.0%
$ 258
COVID Medical Necessity
8.9%
3.9%
50.0%
$ 78
Medical Necessity
4.8%
30.4%
18.4%
$ 553
Out of Network
6.9%
4.4%
2.4%
$ 594
Prior Authorization
0.0%
14.3%
0.0%
$ 421
Underpayment
9.3%
6.9%
6.3%
$ 10
The clinical laboratory segment maintains the lowest volume of denials. But this does not negate the need for robust editing processes. Implementing robust front-end logic and leveraging intelligent automation to correct potential issues dramatically streamlines the process from submission to payment, especially in the high-volume clinical laboratory segment.
% of Total Appeals Filed
% of AppealsPaid after 1st Attempt
% of AppealsPaid after 2nd Attempt
% of AppealsPaid after 3rd Attempt
Avg Paymentper Appeal
Molecular
21.4%
17.2%
19.4%
$1,420
Additional Information
47.7%
23.9%
20.7%
23.3%
$1,285
Medical Necessity
23.0%
17.6%
14.0%
12.8%
$1,518
Prior Authorization
11.4%
18.9%
11.7%
13.1%
$1,944
Experimental and Investigational / Non-Covered
5.6%
13.2%
9.0%
9.0%
$4,234
COVID Underpayment
3.8%
44.7%
24.6%
10.7%
$52
Timely Filing
3.5%
10.1%
8.3%
18.9%
$551
Out of Network
3.5%
14.0%
10.8%
8.4%
$2,513
Underpayment
1.1%
31.2%
17.8%
15.3%
$2,154
COVID Medical Necessity
0.4%
46.4%
27.0%
0.0%
$124
Appeal Trends: Molecular and Genomic Testing At $1,420, the average payment per appeal for molecular testing is more significant due to the high-dollar value of the testing. Additional information appeals account for 47% of the total appeals filed in 2021 in the molecular segment and have an average success rate of 23%. Another 23% of appeals are for claims denied for medical necessity, followed by prior authorizations at 11.4% of total appeals filed. Prior authorization appeal volumes have remained consistent year-over-year in this segment, averaging 10% in 2020, despite a higher volume of prior authorization requirements than pathology or clinical laboratory.
XiFin’s RCM platform has integrated automation with prior authorization partners, allowing claims meeting prior authorization criteria to be submitted to a prior authorization solution automatically. Utilizing “real-time data exchange” via application programming interfaces (API) without partners, XiFin can more quickly acquire the necessary prior authorization number and update the patient’s information in XiFin RPM upon those values being returned.
% of Total Appeals Filed
% of AppealsPaid after 1st Attempt
% of AppealsPaid after 2nd Attempt
% of AppealsPaid after 3rd Attempt
Avg Paymentper Appeal
Pathology
22.6%
20.6%
21.8%
$327
Additional Information
33.4%
28.8%
23.4%
27.9%
$337
Medical Necessity
19.0%
23.5%
23.4%
27.6%
$398
Out of Network
17.9%
17.6%
12.4%
17.7%
$318
Prior Authorization
12.2%
21.5%
32.9%
36.5%
$350
Experimental and Investigational / Non-Covered
9.2%
17.8%
8.9%
3.1%
$195
COVID Underpayment
5.8%
9.0%
3.4%
16.7%
$31
Timely Filing
2.5%
20.5%
15.6%
13.3%
$191
Underpayment
0.1%
52.2%
0.0%
$177
Appeal Trends: Pathology
Approximately 2% of the pathology accessions received into XiFin RPM require an appeal. Those appeals will be responsible for approximately 1-2% of the pathology practice’s revenue. As noted above, the revenue reclaimed is largely attributed to the first attempted appeal. A robust process that includes multiple attempts is critical in revenue recovery in the event the first appeal is not overturned.
If Not Documented, It Did Not Happen
Payer edits and guidelines can be difficult to follow, particularly if physicians, coders, or billing staff are expected to memorize those requirements.
Making the situation even more challenging is the fact that edits vary widely among payers and are constantly changing. RCM platforms should be updated routinely (XiFin RPM is updated monthly) with payer edit updates, while remaining configurable so that custom edits can be easily built to accommodate specific payer requirements.
Whether it is a payer audit or packaging an appeal, documentation in the pathology report and/or clinical notes should clearly outline the services provided and the medical necessity of those services. If it is not documented, it did not happen. Further, understand the various programs that drive payer edits and guidelines. These edits drive an increased need for discipline and documentation. Be conscious of payer-specific requirements. Cigna, Aetna, and UHC require proprietary forms to be completed when appealing claims.
Benchmarking Productivity
Proactively preventing a denial and avoiding the need to submit a corrected claim or file an appeal reduces the time to reimbursement by four to eight weeks, depending on the payer and type of denial. If denials are not addressed properly and manual workflows persist, diagnostic labs will continue to experience a loss of revenue, and staffing will be insufficient to keep up.
Productivity rates for anatomic and molecular billing teams historically average between 12,000-15,000 accessions per person per full-time equivalent (FTE) per year (clinical laboratory is often much higher). However, with the increases in denials, the resulting demands on back-end teams have increased substantially and impacted productivity rates. This holds particularly true for particularly non-covered, medical necessity, and prior authorization denials.
Further, speed to payment is also improved. By automating appeals, the turn-around-time on submitting back to the payer is reduced, on average, from 45 days to 1-3 days, as seen in the blue bar in the chart above.
By installing front-end edits to help maximize clean claims, up to an additional 54 days can be saved, moving from 135 days to just 30 days for full adjudication.
Automating Workflows with AI
Opportunities to automate the process will reduce time and labor and make decisions more consistent. Once there is a deep understanding of coding, billing, denial management, and strategic appeals, there is the ability to automate the important processes across the RCM process. Automation and AI-powered workflows pave the way for consistent, optimized molecular diagnostics and pathology RCM.
Part 3 will demonstrate how AI can be used in RCM to inform, accelerate, automate, validate, and generate.Watch for updates here at DarkDaily.