Clinical laboratories and microbiology tests provide key tools for physicians engaged in antibiotic stewardship programs
One important and continuing trend in healthcare is the need for hospitals, nursing homes, and other medical providers to introduce effective antibiotic stewardship programs (ASPs). The findings of a recent study on antibiotic stewardship emphasize the need for improvement and suggest guidelines that will involve and engage clinical laboratories.
In a recent brief of a study The Pew Charitable Trusts (Pew) conducted with the CDC and various public health and medical experts, Pew wrote, “Minimizing inappropriate antibiotic use in hospitals is a vital element in the fight against antibiotic resistance because more than half of patients admitted to hospitals will receive these drugs. Determining how much antibiotic prescribing is inappropriate and setting national targets to reduce such use are necessary steps for guiding clinical efforts and policies that promote improved antibiotic use.”
To do this, and Pew and the CDC are suggesting “widespread adoption of effective antibiotic stewardship programs, which promote responsible antibiotic prescribing, in order to minimize the harmful effects of inappropriate or unnecessary antibiotic use for patients and slow the spread of resistance.”
And because clinical laboratories perform all the in-hospital testing for ASPs, they will be big part of this effort.
Pew/CDC Set New National Targets for Antibiotic Use Improvement
The Pew brief states that in 2018 the researchers began “to evaluate antibiotic use in hospitals and set national targets to improve prescribing.” The brief adds that “Because of the complexity and diversity of illnesses among hospitalized patients, and the limitations on available clinical data for all antibiotic use in hospitals, the panel focused its analysis on four categories of prescribing that account for the most common antibiotic therapies in US hospitals. Using national prescribing data, the experts examined the use of two types of antibiotics—vancomycin and fluoroquinolones—and antibiotic treatments associated with two conditions: community-acquired pneumonia (CAP) and hospital-acquired urinary tract infection (UTI).”
It their paper published in JAMA Network Open, titled, “Assessment of the Appropriateness of Antimicrobial Use in US Hospitals,” the Pew/CDC researchers wrote, “In this cross-sectional study of 1,566 patients at 192 hospitals, antimicrobial use deviated from recommended practices for 55.9% of patients who received antimicrobials for community-acquired pneumonia or urinary tract infection present at admission or who received fluoroquinolone or intravenous vancomycin treatment.”
Infection Control Today reported that the CDC and Pew set the following goals for hospitals, but did not give a deadline for improvement:
Decrease antibiotic inappropriate prescribing in CAP and UTI cases by 90%.
Decrease overprescribing of fluoroquinolones and vancomycin by 95%.
“Meeting these national reduction targets will require widespread adoption of effective antibiotic stewardship programs, which promote responsible antibiotic prescribing in order to minimize the harmful effects of inappropriate or unnecessary antibiotic use for patients and slow the spread of resistance,” noted the Pew brief, which also pointed out that hospitals should provide incentives to report antibiotic use and impact of stewardship programs to the CDC’s National Healthcare Safety Network (NHSN).
‘Ample Room for Improvement’
The Pew/CDC panel of experts analyzed hospitalized patient data from August 2017 through May 2020. Of those patients, the researchers found that:
219 had CAPs,
452 had UTIs,
550 had received fluoroquinolones, and
403 had received vancomycin.
They also found that:
56% of antibiotic prescriptions were wrong in the type of antibiotic, how long it was used, or why it was chosen.
79% of antibiotic prescriptions for CAP were inappropriate.
77% of antibiotic prescriptions did not suit UTI patients.
47% of fluoroquinolone prescriptions were unsupported.
27% of vancomycin prescriptions were amiss.
The researchers concluded that providers have “ample room for improvement,” the Pew brief notes.
“A substantial percentage of CAP, UTI, fluoroquinolone, and vancomycin treatment was unsupported by medical record data collected (55.9% overall and as high as 79.5% for CAP),” the researchers wrote in their published study.
Pew/CDC Researchers Find Many Antibiotic Prescription Errors
According to the Pew/CDC researchers, missteps in antibiotic usage include:
Treating inpatients too long with antibiotics.
Selecting antimicrobials inconsistent with guidelines.
Absence of signs and symptoms of infection.
Lack of clinical laboratory tests or microbiologic evidence of infection.
The study revealed antibiotic duration errors were most prevalent in the CAP patients, some being treated with antibiotics for more than seven days.
“Almost 60% of the inappropriate prescribing is attributed to exceeding the recommended seven days of treatment, and the use of the wrong antibiotic accounts for most of the remaining inappropriate (CAP) cases,” the Pew brief explained.
Antibiotics Prescribed without Evidence of Infection
As medical laboratory professionals know, microbiology tests identify presence and type of bacteria in urine. But the Pew/CDC researchers reported they found UTI cases that lacked evidence of infection.
“In most instances—where antibiotic use was not supported—the antibiotics were prescribed to patients who lacked symptoms or microbiology test results consistent with UTIs,” according to their report.
Antibiotics Overprescribed to COVID-19 Patients
Another study conducted by The Pew Charitable Trusts “assessed the frequency of bacterial infections and antibiotic prescribing patterns in hospitalized patients diagnosed with COVID-19 in the US.” The researchers, according to the Pew brief on that study, titled, “Could Efforts to Fight the Coronavirus Lead to Overuse of Antibiotics?” used “IBM Watson Health’s electronic health records [EHR] database to capture data about approximately 5,000 patients and nearly 6,000 hospital admissions from February through July 2020.”
The researchers of that study found potential antibiotic misuse among COVID-19 patients as well.
52% received at least one antibiotic prescription.
36% had multiple antibiotics.
96% were treated with antibiotics within 48 hours of admission and likely before infection was confirmed.
“Our data shows that there was very likely a significant amount of unnecessary antibiotic prescribing among hospitalized COVID-19 patients,” Rachel Zetts, Officer, Antibiotic Resistance Project at The Pew Charitable Trusts, told Becker’s Hospital Review. “Overprescribing on this scale could negatively impact the progress we’ve made in the fight against antibiotic resistance over the years, so encouraging physicians to reduce inappropriate antibiotic use and equipping them with the tools needed to do so is critical.” Those tools include test results clinical laboratories produce in support of antibiotic stewardship programs. (Photo copyright: The Pew Charitable Trusts.)
Clinical Laboratories are Key Partners
Hospital-based clinical laboratory leaders may want to contact physicians and infection control colleagues and work toward correcting use of antibiotics in patient care. And microbiologists are advised to aggressively communicate available medical laboratory test data about UTI infections, which the Pew/CDC study suggests can be missed.
Medical laboratories provide testing to diagnose infections and to identify strains of infectious agents that may be antibiotic-resistant. Therefore, lab leaders will be key partners in hospitals’ efforts to reduce infections and prevent antibiotic resistance.
In their letter, the Representatives wrote, “As you are aware, the recently enacted Paycheck Protection Program and Health Care Enhancement Act (PPPHCE Act) invests $25 billion in the [Public Health and Social Services Emergency Fund (PHSSEF)], including $11 billion for states, localities, territories, and tribes, to enhance all aspects of COVID-19 testing capacity. This funding is in addition to the funds already appropriated to the PHSSEF under the CARES Act.
“While laboratories are eligible, along with other providers, for these funds,” they continued, “there have been no federal funds specifically designated for the laboratories that have stepped up in this public health crisis and have made significant investments to expand access to COVID-19 testing despite 40-60 percent reductions in regular commercial volume due to the economic lockdowns.
“As laboratories work to maintain their investments in critical resources for testing platforms, reagents, swabs, and PPE, as well as hiring, training, and overtime pay for the laboratory workforce, we urge HHS to direct a portion of funding that has not already been allocated towards these efforts. These funds will ensure that labs can continue to rapidly scale up diagnostic and antibody testing, particularly for healthcare workers, first responders, and other Americans on the frontlines of this pandemic,” concluded the Representatives.
ACLA President Made Similar Plea for Direct Funding to Clinical Laboratories
“In order to deliver accurate, reliable results for patients at a national scale, we must allocate funding to support [clinical laboratories’] expanded efforts,” she said in a statement following an April 27 meeting at the White House.
In her letter, Khani wrote, “It is essential that HHS allocate $10 billion from the fund to support labs’ further expansion of testing capacity to fulfill the testing needs of all of the states and to protect the lives and livelihood of all Americans.
“Further,” she continued, “HHS should note that investing in the nation’s laboratories will not only enhance testing capacity in the short-term, but it also will allow the country to benefit from a robust testing infrastructure for the duration of the COVID-19 pandemic and beyond.”
President Trump signed H.R.266 into law on April 24. It includes $25 billion earmarked for research, development, validation, manufacturing, purchasing, administering, and expanding capacity for COVID-19 testing. According to the language of H.R.266, that includes, “tests for both active infection and prior exposure, including molecular, antigen, and serological tests, the manufacturing, procurement and distribution of tests, testing equipment and testing supplies, including personal protective equipment needed for administering tests, the development and validation of rapid, molecular point-of-care tests, and other tests, support for workforce, epidemiology, to scale up academic, commercial, public health, and hospital laboratories, to conduct surveillance and contact tracing, support development of COVID-19 testing plans, and other related activities related to COVID-19 testing.”
“As the demand for testing continues to grow, clinical laboratories need dedicated funding to plan for challenges that lie ahead. Strong federal coordination and leadership is essential, and we’re looking forward to working with HHS to ensure that laboratories have the resources necessary to continue to expand our role at the forefront of the nation’s response,” said Julie Khani (above), President of the American Clinical Laboratory Association (ACLA), in a press release following the June 8 letter sent to HHS by 30 members of Congress requesting funds from H.R.266 be sent directly to clinical laboratories. Khani will be speaking on federal policies now impacting clinical laboratories at the upcoming 25th annual Executive War College on Laboratory and Pathology Management in New Orleans on July 14-15. (Photo copyright: ACLA.)
Financial Struggles for Hospitals and Clinical Laboratories
This new round of stimulus funding comes at a time when many providers and clinical laboratories are struggling financially, despite the influx of COVID-19 patients.
“Across the country, laboratories have made significant investments to expand capacity, including purchasing new platforms, retraining staff, and managing the skyrocketing cost of supplies. To continue to make these investments and expand patient access to high-quality testing in every community, laboratories will need designated resources. Without sustainable funding, we cannot achieve sustainable testing,” said Khani in an ACLA statement.
As the COVID-19 coronavirus pandemic evolves, federal regulations, as well as emergency funding for COVID-19 testing that is provided by federal legislation, will evolve in unexpected ways. For that reason, clinical laboratory leaders will want to closely track announcements by such federal agencies as the Department of Health and Human Services, the Centers for Medicare and Medicaid Services, the Food and Drug Administration, the Centers for Disease Control and Prevention, and the Federal Emergency Management Administration as decisions are made about how to assign the $25 billion authorized in H.R.266 for “testing.”
Lack of regulations and quality management jeopardizes the quality and safety of LDTs, claim experts in clinical laboratory medicine in a commentary to Canadian policymakers
The IHPME members published their comments in the Canadian Medical Association Journal (CMAJ), a peer-reviewed journal owned by Joule Inc., a subsidiary of the Canadian Medical Association. In it, they claim “recent expansion of the molecular diagnostics industry has revealed weaknesses in Canada’s regulatory system for laboratory-developed tests, which are not subject to statutory regulations on medical devices.”
For pathologists and clinical laboratory professionals in both Canada and the United States, these recent actions show the concerns many experts have as they watch the explosive growth in the use of laboratory-developed tests in both countries. In many ways, the swift advances in molecular and genetic diagnostics is outrunning the ability of government regulators to keep pace with use of LDTs in clinical care settings.
In their commentary in CMAJ, the IHPME members also
claim the review and evaluation of LDTs in Canada is inconsistent. Some LDTs they
say, may endure stringent assessments and have endorsements by clinical
guidelines or findings that are published in scientific journals. Other LDTs,
however, may have no analysis at all.
In addition, the IHPME members point out that there is no
national registry kept of LDTs. They theorize that a lack of proper regulation,
controls, and quality management “has potentially jeopardized the delivery of
quality, safe, timely, and appropriate care.”
The researchers calling on Health Canada to address these
issues include:
Fiona A. Miller, PhD, Professor of Health Policy and IHPME Chair in Health Management Strategies;
François Rousseau, PhD, Professor, Department of Molecular Biology, Medical Biochemistry and Pathology, Faculty of Medicine, Laval University, Quebec;
Alberto Gutierrez, PhD, Partner, NDA Partners LLC, former Director, Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health (CDRH);
Stuart Hogarth, PhD, Lecturer in Sociology of Science and Technology, University of Cambridge, Cambridge, UK.
During an exclusive presentation offered by The Dark Report (Dark Daily’s sister publication) in 2015, Alberto Gutierrez, PhD (above), who at that time was Director, Office of In Vitro Diagnostics and Radiological Health at the FDA, said, “LDTs are an area that will be difficult to regulate. There is a broad set of tests. Some of the LDTs are very good. Some of them require a lot of expertise from the pathologists and some of them don’t. Regulating LDTs in a way that makes sense and that does not disrupt what’s going on [in clinical laboratories] is going to be difficult.” (Photo copyright: FDA.)
Canadian Scientists Call on Health Canada to Take the
Lead on Regulating LDTs
In the US, the FDA has been making moves to regulate LDTs since 2010, with much opposition from clinical laboratories and In Vitro Diagnostic (IVD) manufacturers. The FDA describes LDTs as internally designed clinical laboratory tests that are developed, manufactured, and used within a single laboratory. They have not undergone government regulatory review, can be simple or complex, and can be utilized to detect a variety of analytes.
Health Canada is the name of a department that falls under
the purview of the Minister of
Health and is part of Canada’s Health
Portfolio. It is responsible for helping Canadians maintain and improve
their health. Other agencies included in the Health Portfolio are:
According to the IHPME paper, however, Health Canada
currently does not have a way to regulate LDTs, and no government agency in
that country is responsible for the oversight of laboratory-developed tests.
Only LDTs that are marketed as test kits are evaluated and reviewed by Health
Canada.
“The current laboratory regulatory system in Canada involves a mixture of public and private entities and operates with oversight from provincial governments, nongovernmental organizations, and professional societies,” the IHPME paper states, adding, “most provinces and territories rely on voluntary standards that are unevenly applied, with little auditing and systematic testing to ensure quality.”
The authors also note that the current lab regulations in
Canada apply only to the operations of the medical laboratories themselves,
encompassing such things as lab environments, personnel, accreditation, and
quality control. They believe the loophole regarding LDTs needs to be addressed,
and they urged Health Canada to “demonstrate leadership” by subjecting these
tests to regulations that are currently applied to medical devices and
pharmaceuticals.
Other Countries Regulate LDTs, though Not Without
Controversy
In support of their call to action, IHPME researchers noted
that Australia, the EU, and the US all have taken steps to regulate LDTs.
The Australian government began oversight of LDTs in 2010 by
subjecting high-risk LDTs to external evaluation and then tracking them in a
public registry.
An EU regulation, which was passed in 2017, will administer
regulatory review of LDTs manufactured on an industrial scale, which targets
commercial laboratories. The law exempts LDTs utilized within individual
hospital laboratories and should be fully implemented by 2022.
Though on its radar since the 1990s, in 2010, the FDA officially
announced its intent to regulate LDTs in the US. The agency released an initial
draft approach for doing so starting in 2014, held a public workshop on the
topic in 2015, and released a
discussion paper in 2017. At this time, however, the FDA is not regulating
LDTs, though the agency remains open to the possibility.
Dark Daily
has reported extensively over the years on the development of LDTs and the
controversy surrounding the FDA’s moves to regulate them.
According to the FDA
website, problems with several high-risk LDTs have been identified,
including:
Claims that are not adequately supported with
evidence;
Lack of appropriate controls which may yield
erroneous results; and
The FDA’s report, titled, “The
Public Health Evidence for FDA Oversight of Laboratory Developed Tests,” reviewed
20 case studies of LDTs for Lyme disease, ovarian cancer, whooping cough,
fibromyalgia, prostate cancer, autism, breast cancer, melanoma, Vitamin D, and
other conditions. The agency concluded that in many instances “patients have
been demonstrably harmed or may have been harmed by tests that did not meet FDA
requirements.”
Klein noted, however, that “The 20 tests described by FDA are mostly a hodgepodge of outlier assays including tests that were never offered, tests for which comparable FDA assays perform poorly, tests for poorly defined disorders with psychologic components, and use of an FDA-approved test off-label.” He continued, “That FDA could find only these dubious examples out of the many thousands of laboratory-developed procedures (LDPs) that benefit patients each day, calls into question the agency’s rationale for expanding its regulatory scope to include LDPs.”
Perhaps this is why the FDA has yet to implement regulations
for LDTs. The controversy continues.
Whether Health Canada will accept the advice of the IHPME
scientists and take steps to regulate laboratory-developed tests in Canada remains
to be seen. As more LDTs are created and manufactured, however, it is probable
that governments will continue to evaluate the administration and oversight of laboratory-developed
tests.
In both Canada and the United States, pathologists, clinical
laboratory managers, and executives at in vitro diagnostic manufacturers
can expect an ongoing tug-of-war between government regulators and the lab
industry over the most appropriate ways to regulate LDTs.
This CMS pilot program is another opportunity for clinical laboratories to provide medical lab test services and collect specimens outside of traditional sites of healthcare services
Clinical laboratories and anatomic pathology groups are once again reminded to develop strategies that support the increasing number of physicians providing medical care in nontraditional outpatient settings. Now in its seventh year, the Medicare Independence at Home program is reviving the tradition of healthcare providers making house calls to elderly patients who have certain chronic illnesses, and so far, the results are promising.
Primary care teams at the 14 participating healthcare
providers include physicians, nurse practitioners, physician assistants,
pharmacists, social workers, and other staff.
Hospital networks participating in the federal Centers for Medicare and Medicaid Services (CMS) primary care pilot program are saving the government millions of dollars, while improving healthcare outcomes for their chronically ill patients and earning millions in return.
A CMS fact sheet states that to qualify for incentive payments, participating providers must meet performance thresholds of at least three of the following six measures:
Follow-up contact within 48 hours of a hospital
admission, hospital discharge, and emergency department visit;
Medication reconciliation in the home within 48
hours of a hospital discharge and emergency department visit;
Annual documentation of patient preferences;
Hospital admissions for ambulatory care
sensitive conditions; and
Emergency department visits for ambulatory care
sensitive conditions.
Northwell Health House Calls a Model of Success
The Independence at Home (IAH) demonstration project from the federal Center for Medicare and Medicaid Innovation (CMMI) was established in 2010 as part of the Affordable Care Act. In 2018, Congress extended the pilot for another two years and increased the number of eligible participants from 10,000 to 15,000.
Northwell Health House Calls has been a model of success within the federal IAH demonstration project. The New York-based healthcare provider has annually reduced costs while improving health outcomes for participating patients.
Karen Abrashkin, MD (above), Medical Director of Northwell Health House Calls, examines a patient during a home visit checkup. In a news release, she said, “We know our older, chronically ill patients want to receive medical care at home as long as possible. Programs like Independence at Home involve a large interdisciplinary team working in concert to deliver individualized patient care. We are dedicated to providing high-quality care and giving patients access to the appropriate healthcare provided at the right time.” (Photo copyright: Northwell Health.)
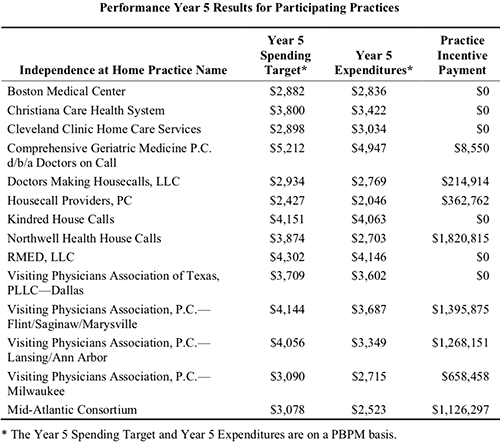
Results from the fifth year of the program (Oct. 1, 2016 through Sept. 30, 2017) show Northwell Health reduced per-beneficiary-per-month (PBPM) expenditures to $2,703, compared to a spending target of $3,874, according to the most recent CMS Fact Sheet. In return, Northwell Health received an incentive payment of more than $1.82 million. That’s the largest payout among the eight practices that met incentive payment quality benchmarks and savings requirements.
According to the news release, patients in Northwell’s House Calls program receive comprehensive, coordinated care, that includes ultrasounds, radiology, electrocardiograms, sleep studies, clinical laboratory work, physical exams, occupational and speech therapy, and social services, as well as intravenous fluids and prescription refills.
Physicians, nurse practitioners, and other clinicians are
available for urgent, same-day visits during the work week. The team also is
accessible 24/7 to answer clinical questions from patients and caregivers, or
to arrange urgent services.
In an interview with Crain’s New York Business, Karen Abrashkin, MD, Medical Director of Northwell Health House Calls, said, “We’ve achieved cost savings by providing really good primary care and ongoing care for medical illnesses. We’re responsive to patients whenever they have a change in condition.”
The chart above is taken from the federal Independence at Home (IAH) Year Five Fact Sheet, released October 25 of this year. CMS found that “the actual expenditures for IAH practices’ applicable beneficiaries were approximately 8.4% (equating to $33.5 million) below their spending targets, an average reduction of $2,711 per beneficiary. Thirteen out of the 14 IAH practices reduced the per-beneficiary-per-month (PBPM) expenditures relative to the practice’s PBPM spending target. (Chart copyright: Centers for Medicare and Medicaid Services.)
How Patients Qualify for Medicare’s IAH Program
To qualify for the Independence at Home pilot, patients must:
Currently be Medicare beneficiaries with two or
more chronic health conditions;
Need help with activities of daily living; and
Have had a hospital admission and rehab stay
within the past year.
Though he praises the House Calls program’s success, Kristofer Smith, MD, Senior Vice President of Population Health Management at Northwell Health stated that the program should be expanded slowly and only extended to those who would benefit most from in-home care.
“We need to be thoughtful about making sure we’re not expanding beyond the populations for whom we know it works because [it would] dilute the results,” he told Modern Healthcare.
US Congressman Michael C. Burgess, MD, (R-Texas), said in a statement last July announcing a proposed bill to make the program permanent, “The Independence at Home program is a fiscally-responsible solution to help seniors access quality healthcare and expand the capacity of our nation’s healthcare system. Under this program, high-needs patients continue to receive individual care in the comfort of their homes, reducing unnecessary hospitalizations and allowing physicians and primary care teams to spend more time with patients.” [Photo copyright: US Congress.]
Will Medicare’s Primary Care at Home Program Continue
Beyond the Pilot?
The Independence
at Home pilot is scheduled to end Dec. 31, 2020. What happens next is
uncertain. Efforts in Congress to create a permanent home-based primary care
program under Medicare have not yet gained traction despite bipartisan support.
Thomas Cornwell, MD, CEO, Home Centered Care Institute (HCCI), a national non-profit organization focused on advancing home-based primary care, is skeptical the primary care provider workforce could meet increased demand. He told Home Health Care News that question is “the greatest unknown.”
Nevertheless, the apparent success of Medicare’s
Independence at Home pilot program should be a wakeup call to clinical
laboratories and anatomic pathology groups that the trend of providing medical
services in lower-cost settings will likely continue.
That means medical laboratory leaders should be developing
strategies to support providers who are delivering medical care in nontraditional
healthcare environments.
SIDM estimates approximately one in 10 patients with serious medical conditions are initially misdiagnosed, a problem that can be addressed if the right medical laboratory test is ordered at the right time for individual patients
Clinical
laboratory leaders know that lab tests are essential to a large proportion
of medical diagnoses. Therefore, any formal effort to reduce diagnostic errors that
affects how doctors order and use lab tests also will impact medical
laboratories that perform those tests.
“Diagnostic error is one of the most important safety
problems in healthcare today and causes the most patient harm,” said Paul L. Epner, CEO and
co-founder of the SIDM, in a news
release. “While many organizations have diagnostic quality on their radar,
it generally is not seen as a top priority. Those who’ve joined the coalition
acknowledge that diagnostic quality and safety are vital to improving
healthcare.”
To participate in the coalition, organizations must promise
to pursue ways to circumvent troublesome diagnostic errors and submit action
plans to the SIDM outlining proposals to diminish such errors. There are no
fees associated with being part of the coalition.
12-Million Patients Each Year Affected by Diagnostic
Errors
Epner told Modern
Healthcare that this coalition is the only one that exists that focuses
solely on diagnostic errors and ways to eradicate them. “There are a lot of
systematic things that we understand are problems, but we aren’t very good at
implementing solutions,” he said. “In terms of having standard solutions, we
are early.”
The National
Academy of Medicine (NAM) defines diagnostic error as “the failure to
establish an accurate and timely explanation of the patient’s health problem(s)
or to communicate that explanation to the patient. Simply put, these are
diagnoses that are missed altogether, wrong, or should have been made much
earlier.”
The SIDM website lists the following key sources for
acquiring data on diagnostic errors:
Autopsy data;
Physician self-reports of experiencing
diagnostic error;
Patient self-reports of experiencing diagnostic
error;
Hospital incident reporting systems;
Statistical analysis of unexpected
hospitalizations;
Research studies designed to measure diagnostic
error; and
Medical malpractice claims data.
“Providing an accurate medical diagnosis is complex and involves uncertainty, but it’s obviously essential to effective and timely treatment,” Paul L. Epner, CEO and co-founder of the SIDM, said in a news release announcing the formation of the coalition. “Nearly everyone will receive an inaccurate diagnosis at some point in their life and for some, the consequences will be grave. Major improvement is needed to systematically identify how to improve diagnostic quality and reduce harm to patients.” (Photo copyright: Society to Improve Diagnosis in Medicine.)
The SIDM
states that diagnostic errors affect an estimated 12-million patients in
the US each year, and that approximately one in 10 patients with a serious
medical condition are initially misdiagnosed. In addition, an estimated 40,000
to 80,000 people die each year from diagnostic errors in US hospitals, and it
is probable that at least that many patients suffer from permanent disability
annually due to improper diagnosis. It is also likely that diagnostic errors
cause more harm to patients than all other medical errors combined and are
responsible for an increasing number of malpractice cases, the SIDM notes.
John’s Hopkins Finds Most Misdiagnoses in Three
Categories of Medicine
A study published in the peer-reviewed journal Diagnosis,
reported that one in three malpractice cases that resulted in death or serious
harm to patients are due to misdiagnosis. The research for this study was
carried out by a team from the Johns Hopkins University
School of Medicine and was funded by the SIDM.
After analyzing more than 55,000 malpractice claims, the
researchers found that 34% of those cases which resulted in death or permanent
disability could be attributed to inaccurate or delayed diagnosis, an SIDM
analysis of the John’s Hopkins study noted.
The research team also examined underlying disease states to
search for misdiagnosis patterns and discovered that three quarters or 74.1% of
the misdiagnosed cases occurred in just three categories of medical conditions:
Cancer (37.8%);
Vascular events (22.8%); and
Infection (13.5%).
These serious cases resulted in $1.8 billion in malpractice
payouts over the course of 10 years, according to the SIDM.
“It is not just inconvenient to have a wrong or delayed
diagnosis. For many patients, misdiagnosis causes severe harm and expense, and
in the worst cases, death,” said David
Newman-Toker, MD, PhD, Director, Armstrong
Institute Center for Diagnostic Excellence at Johns Hopkins University
School of Medicine, in an SIDM
news post. “If we’re going to reduce serious harms from medical errors,
major strides must be made to improve diagnostic accuracy and timeliness. This
study shows us where to focus to start making a difference for patients. It
tells us that tackling diagnosis in these three specific disease areas could
have a major impact on reducing misdiagnosis-related harms.”
The John’s Hopkins research confirms that misdiagnosis is a
common and costly form of medical errors that can have catastrophic results.
The team concluded that it will take a system-wide effort involving physicians,
patients, and their families to improve the accuracy of diagnosis.
The SIDM’s Coalition to Improve Diagnostics is one such
effort and is primarily supported by a $2.45 million grant awarded by the Gordon and Betty Moore Foundation. The main
purpose for this grant was to help increase awareness about diagnostic errors
and develop ways to prevent such errors in the future.
“We think this is a new frontier of safety and quality we
want to be part of,” Daniel
Yang, MD, Program Officer for Diagnostic Excellence Initiative at the Moore
Foundation, told Modern Healthcare.
Clinical laboratory tests are essential to the diagnostic
process. Therefore, lab managers and staff should constantly review their procedures
to ensure accuracy in testing and reporting of results to ordering physicians.
If preventable medical errors are to be significantly reduced, labs will be a
big part of the team effort that will make it happen.
Pharmaceutical developers are combining genetic sequencing and precision medicine to create new drug therapies and cancer treatments designed for specific patients
Most anatomic pathologists are aware of the rapid advances in the field of cancer immunotherapy—sometimes also called immune-oncology. This is an area of healthcare where precision medicine and personal genetics become crucial elements in developing more effective drug regimens.
Scientists are combining those two areas of research to develop vaccines designed for specific individuals based on the genetic characteristics of their DNA. This is why there are great hopes that cancer immunotherapy can be used to artificially stimulate the immune system to treat cancer and improve the system’s natural ability to fight cancer.
San Francisco-based Genentech, a subsidiary of Swiss pharmaceutical giant Roche (OTCMKTS:RHHBY), is working with German company BioNTech to develop such personalized vaccines for cancer patients. Each vaccine would be based on the unique deoxyribonucleic acid (DNA) of a patient’s tumor.
Unlike typical vaccines, Genentech’s drug would not be taken
as a preventative measure. Instead, patients receive it after being diagnosed
with cancer.
Though
still being tested, this new line of research indicates that development of personalized
cancer treatments is progressing, as scientists strive to customizetreatments tumor by tumor.
Creating One-Off Vaccines
To create each vaccine, a patient first undergoes a tumor biopsy. The sample tissue is then sent to a genetics laboratory for full genome sequencing. Sophisticated algorithms analyze the genetic data and locate targets within the tumor that have the most potential for training the patient’s immune system to attack the existing cancer. A customized vaccine is then created for and administered to the patient.
“What’s truly revolutionary about this approach is that each vaccine uses a common molecular backbone—mRNA—that is uniquely tailored to an individual patient,” said Todd Renshaw, former Global Head of Clinical Contract Manufacturing at Genentech, in an article posted on the company’s website. “It’s the next step in personalized medicine.”
Vaccines are typically used to train the body’s immune
system to attack specific diseases that infiltrate the body from the outside. However,
cancer tumors are formed within the body’s own tissues, making it difficult for
the immune system to detect them. Thus, vaccines haven’t shown much promise for
treating cancer.
“Vaccines work by exposing the immune system to ‘non-self’ proteins known as antigens, priming it to recognize and eliminate the invaders. But in the case of cancer cells, most proteins are the same as those on healthy cells,” said Lélia Delamarre, Senior Scientist in Cancer Immunology at Genentech, in the online article. “This makes it hard to identify which antigen to use in a vaccine.”
Global
testing on the vaccine has commenced with a focus on ten cancers in upwards of
560 patients.
Barriers to Creating Individual Vaccines
The American Cancer Society estimates there were 1,735,350 new cancer diagnoses in the US in 2018—and 609,640 cancer deaths—making it the second leading cause of death in the US after heart disease.
A
truly customized cancer treatment in the form of a vaccine could be a major
breakthrough in treating this deadly disease. However, there are significant
barriers to developing such a vaccine.
For
starters, the vaccines cannot be manufactured in batches, packaged, warehoused,
or delivered to pharmacies in bulk. The personalized vaccines must be
manufactured in single patient doses, which could be prohibitively costly.
Nevertheless, this research represents an exciting
opportunity for anatomic pathologists and clinical laboratories with genetics
capabilities which would be needed to secure and sequence tumor biopsies for
guiding the creating of the customized vaccines.
Pathologists should track this trend closely and work within
their group practices to ensure they have the analyzers, informatics, and
expertise required to perform this type of testing for patients within their
communities.