Federal regulators continue to recognize value of clinical laboratory testing in near-patient settings
To help in the diagnosis and management of two sexually-transmitted diseases, another point-of-care diagnostic test will soon be available for use in physician’s offices, urgent care clinics, and other healthcare settings. The federal Food and Drug Administration (FDA) announced it granted a CLIA waiver for the binx health io CT/NG assay, a molecular platform used to detect sexually transmitted diseases—chlamydia and gonorrhea—at the point of care (POC).
This will be welcome news to many medical professionals, as it indicates federal regulators recognize the value of diagnostic testing in near-patient settings.
Allows Non-Laboratorian Processing at Point of Care
In 2019, binx health received FDA 510k clearance to market its binx io rapid point-of-care (POC) platform for women’s health. “The binx io platform is a rapid, qualitative, fully-automated test, designed to be easy to use, and intended for use in POC or clinical laboratory settings … In the company’s recently completed 1,523-person, multi-center clinical study, 96% of patient samples were processed on the binx io by non-laboratorians in a POC setting,” a binx press release noted.
According to the Boston-based biotech company’s website, the binx io platform (above) combines ultra-rapid, polymerase chain reaction (PCR) amplification with binx health’s proprietary and highly sensitive electrochemical detection technology. The io instrument processes a single-use, CT/NG cartridge that contains all reagents for testing self- or clinician-collected vaginal swabs and male urine samples. No sample preparation is required. Test results are available in less than 30 minutes. (Photo copyright: binx health.)
“With ever-increasing sexually transmitted infection rates, point-of-care and CLIA-waived platforms like the binx io are essential additions to our sexually-transmitted-infection-control toolbox, which will increase accessibility and decrease the burden on traditional healthcare settings,” Barbara Van Der Pol, PhD, Professor of Medicine and Public Health at University of Alabama at Birmingham, said in a binx press release.
According to binx, the Centers for Disease Control and Prevention (CDC) estimates that one in five people in the US has a sexually-transmitted disease (STD), with an estimated 108 million Americans potentially in need of routine STD testing. Additionally, chlamydia and gonorrhea are the two most treated STDs globally.
Study Finds Binx Health POC Assay Comparable to Traditional Clinical Laboratory NAATs
Van Der Pol led a team of researchers who compared the binx io chlamydia/gonorrhea POC assay to three commercially-available nucleic acid amplification tests (NAATs). The binx-funded study, published in JAMA Network Open, analyzed swab samples from 1,523 women (53.6% with symptoms) and urine samples from 922 men (33.4% symptomatic) who presented to 11 clinics in nine cities across the US.
The molecular point-of-care assay proved on par with laboratory-based molecular diagnostics for vaginal swab samples, while male urine samples were associated with “good performance.”
For chlamydia:
Sensitivity of the new POC assay was 96.1% (95% CI, 91.2%-98.3%) for women and 92.5% (95% CI, 86.4%-96.0%) for men.
Specificity of the new POC assay was 99.1% (95% CI, 98.4%-99.5%) for women and 99.3% (95% CI, 98.4%-99.7%) for men.
For gonorrhea:
Sensitivity estimates were 100.0% (95% CI, 92.1%-100.0%) for women and 97.3% (95% CI, 90.7%-99.3%) for men.
Specificity estimates were 99.9% (95% CI, 99.5%-100%) for women and 100% (95% CI, 95.5%-100%) for men.
Van Der Pol told Reuters News, “The bottom line is that chlamydia and gonorrhea are still the most frequently reported notifiable diseases in the US, and it costs us in the $5 billion to $6 billion range to manage the consequences of untreated infections. Unfortunately, about 70% of women who are infected don’t have any symptoms, so they don’t know they need to be tested.”
“The ability to diagnose at a point-of-care setting will help with more quickly and appropriately treating sexually-transmitted infections, which is a major milestone in helping patients,” said Tim Stenzel, MD, PhD (above), Director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health, in the FDA announcement. “More convenient testing with quicker results can help patients get access to the most appropriate treatment. According to the CDC, one in five Americans are diagnosed with sexually-transmitted infections every year, which is why access to faster diagnostic results and faster, more appropriate treatments will make significant strides in combatting these infections,” he added. As point-of-care testing for specific diseases increases, clinical laboratories that process these tests may see a decrease in specimen processing orders. (Photo copyright: Duke University.)
The CLIA waiver allows binx to distribute the chlamydia/gonorrhea test to 220,000 CLIA-waived locations across the US through the company’s national commercial distribution partnership with McKesson. Obstetrician/gynecologist and primary care offices, urgent care facilities, community health clinics, STD clinics, and retail settings are all potential testing sites.
Binx says its testing platform can improve health outcomes by:
Increasing treatment compliance,
Limiting onward transmission,
Minimizing the risk of untreated conditions, and
Ensuring the right treatment is provided.
In the binx health press release, binx CEO Jeffrey Luber, JD, said, “The io instrument’s demonstrated clinical effectiveness, ease of operation, and patient convenience make it a much-needed tool with transformative implications for public health, especially now during the COVID-19 pandemic, where STI [sexually-transmitted infection] prevention services nationwide have been dramatically reduced or cut altogether as resources have been allocated to focus on the COVID response.”
Should Clinical Laboratories Be Concerned about POCT?
It happens often: after consulting with his or her doctor, a patient visits a clinical laboratory and leaves a specimen. The test results arrive at the doctor’s office in a few days, but the patient never returns for treatment. That is why point-of-care tests (POCTs) came to be developed in the first place. With the patient in the clinic, a positive test result means treatment can begin immediately.
As the US healthcare system continues toward more integration of care and reimbursement based on value, rather than fee-for-service, point-of-care testing enables physicians and other healthcare providers to diagnose, test, and prescribe treatment all in one visit.
Thus, it is a positive step for healthcare providers. However, clinical laboratories may view the FDA’s increasing endorsement of waived point-of-care testing as a trend that is unfavorable because it diverts specimens away from central laboratories.
There also are critics within the medical laboratory profession who point out that waived tests—often performed by individuals with little or no training in laboratory medicine—have much greater potential for an inaccurate or unreliable result, when compared to the same assay run in a complex, CLIA-certified clinical laboratory.
Dozens of Chicago-area schools were reopened with the help of an $11 COVID-19 saliva test, but the qualifications of the clinical laboratory, and whether it complied with federal regulations, were called into question
It was only a matter of time when newly-formed clinical laboratories—taking advantage of the federal government’s loosening of regulations to promote COVID-19 testing—drew the attention of state regulators and the national news media. This is what happened at New Trier High School in Winnetka, Ill.
In March, the New York Times published an article, titled, “Why Virus Tests at One Elite School Ran Afoul of Regulators.” The article highlighted the coronavirus screening program implemented at New Trier High School and suggested that “New Trier may have inadvertently violated federal regulations on testing,” adding that “the Illinois Department of Public Health (IDPH) opened an investigation into the lab.”
SafeGuard Surveillance of Brookfield, Ill., was contracted to perform the routine saliva-based testing. SafeGuard analyzed saliva samples from students, teachers, and school staff to detect the presence of the SARS-CoV-2 coronavirus. New Trier was just one of several school districts that contracted with SafeGuard for the testing, which costs $11 per test. The samples were typically processed the same day.
“This has been a really valuable safety mitigation for our district to make our staff, students, and community feel safer,” Chris McClain, Assistant Superintendent for Finance and Operations at Glenbard High School District 87, told the Chicago Tribune. “We’ve been very pleased with the program.” Glenbard also contracted with SafeGuard for the COVID-19 surveillance screening.
COVID-19 Surveillance or Screening?
Though the surveillance screening testing was working as intended for multiple Chicago areas school systems, the New York Times article called into question whether SafeGuard—which at the time lacked CLIA (Clinical Laboratory Improvement Amendments) certification—was qualified to conduct COVID-19 screening testing.
The article also alleged that SafeGuard was led by a scientist who was not qualified under the federal guidelines to run a diagnostic laboratory, and that the saliva test being used was not authorized for COVID-19 testing by the federal Food and Drug Administration (FDA).
It came down to whether SafeGuard was conducting “surveillance” testing, which does not require CLIA-certification, or “screening” which does.
SafeGuard was founded by Edward Campbell, PhD, Assistant Professor in the Department of Microbiology and Immunology at Loyola University in Chicago. Campbell, a virologist with decades of experience developing tests for HIV, “adapted a saliva-based coronavirus test last summer and first established a [COVID-19] lab for the suburban school district where he serves on the board,” Patch News reported.
Microbiologist Edward M. Campbell, PhD (above), founded SafeGuard Surveillance toward the end of 2020 after demand for COVID-19 screening he had been conducting for various local school systems increased dramatically. In January, the startup clinical laboratory was running about 25,000 tests per week, the Riverside/Brookfield Landmark reported. (Photo copyright: Loyola University.)
SafeGuard Claims It Complied with Federal Regulations
SafeGuard’s COVID-19 screening tool utilizes RT-LAMP (reverse transcription loop-mediated isothermal amplification) to look for the SARS-CoV-2 coronavirus in saliva samples. This test is less sensitive than the more commonly used polymerase chain reaction (PCR) test that uses a nasal swab to detect the virus. However, the RT-LAMP test is considered reliable, particularly in individuals with a high viral load. The RT-LAMP test also is less expensive than the PCR test, which makes it appealing for public school systems.
To use the RT-LAMP test, faculty, staff, and students spit into test tubes at home and then take the sample to their school or other drop-off location. Campbell’s lab then processes the samples.
After the New York Times article came out, both New Trier and SafeGuard denied they had done anything wrong, and that their screening program complied with government regulations for COVID-19 testing. Campbell maintained that he did not need the CLIA certification to operate his lab for testing and that SafeGuard complied with all federal regulations. Nevertheless, in March, SafeGuard applied for and received CLIA-certification to “conduct ‘screening’ testing, instead of just ‘surveillance’ testing,” Patch News reported.
“We’re doing everything we can to operate in good faith under the guidance that clearly exists,” Campbell told The Chicago Tribune.
In a statement, New Trier district officials said, “New Trier has also met with local and state health authorities to review our use of the program and they have not directed us to change our use of it. From the time the program began, New Trier has been clear that the saliva program is non-diagnostic and must be confirmed by a lab test. To suggest otherwise is false,” Patch News reported.
Surveillance Testing versus Screening
In August, the federal Centers for Medicare and Medicaid Services (CMS), which oversees CLIA labs, released guidelines that stated COVID-19 testing could be performed in clinical laboratories that were not CLIA-certified so long as patient-specific results are not reported.
This “surveillance testing” is intended to identify the disease within a population group and not diagnose individuals. If a person tests positive for COVID-19 via SafeGuard’s saliva test, the individual is directed to get an FDA-approved test to confirm the diagnosis.
“We do definitely see the value of surveillance testing and how that can be used to help schools make informed decisions about remote, in-person, or hybrid learning,” Melaney Arnold, State Public Information Officer for the Illinois Department of Public Health (IDPH) told the Chicago Tribune. She added that the IDPH wants to provide schools with the tools they need to navigate the pandemic.
Following the New York Times article about New Trier High School and SafeGuard’s COVID-19 screening program, the Illinois Department of Public Health opened an investigation into the company. However, the investigation has ended, and the state agency is not taking any further action against SafeGuard, Patch News reported.
It’s worth noting that it was the FDA’s relaxing of federal regulations that encouraged the development of startup clinical laboratories like SafeGuard in the first place. There is, apparently, a fine line between surveillance and screening, and clinical laboratories engaged in one or the other should confirm they have the required certifications.
Former CEO Elizabeth Holmes now awaits March 9 court date on federal fraud charges that include reporting false medical laboratory test results on some patients
Clinical laboratory leaders have watched with keen interest the federal criminal proceedings against disgraced Theranos founder and former CEO Elizabeth Holmes, whose blood-testing company lost nearly $1 billion of investors’ money before dissolving in 2018.
In a recent CNBC interview, John Carreyrou, the Wall Street Journal (WSJ) investigative journalist who first broke the Theranos story in 2015, contended that the once-high-flying Silicon Valley startup could have paid back investors on a pro-rata basis, but that the company opted to use its dwindling cash to challenge lawsuits.
“If you rewind to October 2015, when I finished, when I published my first investigative story on Theranos, the company still had $400 million in the bank and it could have called it quits then,” Carreyrou said in the interview. “And Elizabeth Holmes could have apologized to investors, to patients, to everyone she had misled and returned that money to shareholders on a pro-rata basis.”
Theranos Scandal Breaks Wide Open
Carreyrou’s nearly year-long Wall Street Journal investigation into Theranos helped bring down the venture capital darling that had achieved a $9 billion private valuation before crumbling under the weight of fraud allegations. Dark Daily and our sister publication The Dark Report (TDR) covered in detail the allegations against and investigation into the embattled blood test company in dozens of e-briefings and TDR articles starting in 2015.
In fact, The Dark Report was first to publish the news that Theranos had ceased using its finger-stick collection method in Phoenix as early as April 2015. (See TDR, “Theranos: Many Questions, But Very Few Answers,” April 20, 2015.) At that time, Theranos declined to respond to The Dark Report’s requests for comments.
Theranos had built its superstar reputation on the backs of a revolutionary finger-prick blood testing system, which Holmes promised could diagnosis diseases ranging from diabetes to cancer with just a few drops of blood. But an in-depth investigation into hoopla surrounding the company’s breakthrough technology by Carreyrou and other reporters at the Wall Street Journal revealed it was based on false test results and phony claims to investors and companies, such as Walgreens, which had planned to feature the technology in their retail clinics.
Elizabeth Holmes (above), founder and former CEO of now defunct Theranos, was considered a wunderkind when, as a 19-year-old Stanford University dropout, she founded Theranos in 2003. Early on, she attracted high-profile members to the Theranos board, including former US Secretary of State George Schultz, and cultivated comparisons to legendary Apple CEO Steve Jobs. But once the accuracy of Theranos’ capillary blood-test device fell under suspicion, Holmes’ fall from grace was swift, as clinical laboratories learned from multiple Dark Daily e-briefings and articles in The Dark Report going back to 2015. (Photo copyright: The New York Times.)
In 2016, Theranos received sanctions from the Centers for Medicare and Medicaid Services (CMS), which included revocation of the company’s CLIA certificate and sanctions against Holmes and other company officials that prohibited them from owning or operating a medical laboratory for two years. Soon afterward, Theranos laid off 340 workers, closed its laboratory operations, and shuttered its wellness centers to “focus on an initiative to create miniature medical testing machines,” the New York Times reported.
When Theranos was finally dissolved in September 2018, Carreyrou reported that the company had an estimated $5 million in cash to distribute to unsecured creditors. All told, Carreyrou estimates Theranos’ investors, which included such big names as News Corp Executive Chairman Rupert Murdoch, Bechtel Group Chairman Riley Bechtel, and US Education Secretary Betsy DeVos, lost nearly $1 billion.
Today, Holmes is preparing to stand trial on a dozen federal wire fraud and conspiracy to commit wire fraud charges at the US District Court in San Jose, Calif., where jury selection is slated to start on March 9, 2021, amid COVID-19 pandemic safety precautions.
According to the Mercury News, Holmes faces maximum penalties of 20 years in prison and a $2.75 million fine, plus possible restitution. Carreyrou does not expect Holmes to seek a plea deal.
“I think that the chances of that are pretty unlikely. From what I hear, she’s telling her friends and her entourage that she’s actually looking forward to her day in court and she thinks that the real story—her version of the story—will come out at trial,” he told CNBC. “And so, she’s actually putting on a cheerful face with people she knows, and people have seen her recently and are saying that she’s looking forward to see this go to a jury.”
While the final chapter of this story will be written by a federal court jury, clinical laboratory leaders likely will want Holmes to face maximum penalties if found guilty of all charges. The deceptive scientific and business practices Theranos allegedly engaged in caused many headaches for the clinical lab directors of hospitals and health networks as their CEOs asked why the “cheap and fast” Theranos testing system could not be used instead of traditional, more expensive testing methods.
Theranos also financially damaged investors who might otherwise have gained capital and continued to invest in more credible startups of diagnostic companies and clinical laboratories.
If the proposed rule becomes final, it may shift some inpatient medical laboratory testing away from hospital labs and to independent clinical laboratories
Medical laboratories in hospitals and health systems already feel the pinch of less test orders originating from their own emergency departments (ED). Now, more tests associated with inpatient care might also shift away from hospital labs due to a new proposed rule from the federal Centers for Medicaid and Medicare Services (CMS) that would move 1,740 specific procedures from inpatient care settings to outpatient ambulatory surgical centers (ACS).
Further, the proposed rule would completely phase out the “inpatient only” (IPO) list of services over a three-year transitional period, with total elimination of the IPO list by Calendar Year (CY) 2024.
If finalized as written, the rule (CMS-1736-P) would have a negative impact on the finances of hospitals laboratories as more patients get their care in outpatient settings instead of their local hospitals.
Conversely, hospital outreach labs that service ambulatory surgical centers and other outpatient settings could have an opportunity to pick up more medical laboratory test referrals.
The proposed rule, titled, “Medicare Program: Hospital Outpatient Prospective Payment and Ambulatory Surgical Center Payment Systems and Quality Reporting Programs; New Categories for Hospital Outpatient Department Prior Authorization Process; Clinical Laboratory Fee Schedule: Laboratory Date of Service Policy; Overall Hospital Quality Star Rating Methodology; and Physician-Owned Hospitals,” was published in the Federal Register on August 12, 2020, and is open for comments until 10/05/2020.
“In this proposed rule, we describe the proposed changes to the amounts and factors used to determine the payment rates for Medicare services paid under the OPPS and those paid under the ASC payment system.
Moving from Highest Cost Settings to Lower Cost Settings
In the big picture, these changes can save Medicare money. By shifting procedures for Medicare patients from the highest cost settings—hospital inpatient—to lower cost settings, such as outpatient ambulatory surgical centers, and by eliminating the inpatient-only list, physicians have more leeway to determine for themselves whether a patient needs to be hospitalized for any given procedure.
In “Do Hospitals Have a Target on their Back?” healthcare coding and reimbursement consultant, Terry Fletcher, an editorial board member with ICD10monitor, wrote, “Last year, CMS proposed removing certain services from the inpatient-only list and making them available on an outpatient basis, which it said would help lower costs.
“According to the proposal, ambulatory surgical centers would get a payment increase of 2.6%, and CMS estimated total payments to them for 2021 will be about $5.45 billion, an increase of $160 million from this year,” she added.
Fewer Referrals for Inpatient Lab, More for Hospital Outreach Labs
The impact of the proposed rule is predictable—price shopping will ensue, which is what Medicare wants. Thus, with the removal of the inpatient-only procedure list, the clinical laboratories of hospitals and health systems will likely see a reduction in inpatient test orders. But clinical laboratories participating in hospital outreach programs may see an increase in test orders, as doctors transition to more outpatient procedures.
This seemingly simple shift may be more complicated than it appears, however, for both patients and labs. “In general, any routine test is going to be more expensive at a hospital,” Jean Pinder, founder and CEO of ClearHealthCosts, told Cleveland.com.
There may be other concerns as well. Convenience, insurance coverage, and physician recommendations often influence patient decisions about clinical laboratories.
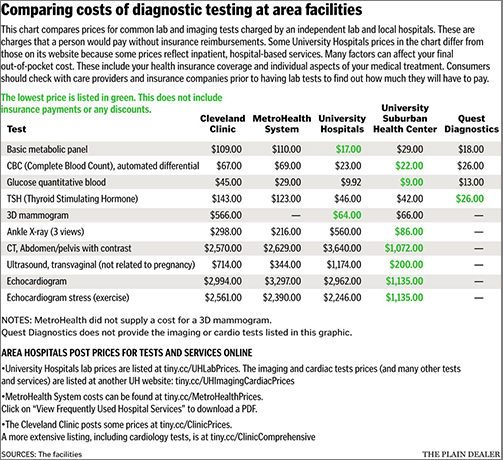
The chart above, taken from the article by Cleveland.com, “compares prices for common clinical laboratory and imaging tests charged by an independent lab and local hospitals. These are charges that a person would pay without insurance reimbursements. Some University Hospitals prices in the chart differ from those on its website because some prices reflect inpatient, hospital-based services. Many factors can affect [a patient’s] final out-of-pocket costs, including health insurance coverage and individual aspects of your medical treatment.” (Graphic and caption copyright: Cleveland.com.)
Change Is the Only Constant
The entire healthcare industry is undergoing change that is unlikely to end any time soon. Clinical laboratory managers who stay aware of trends in the industry and remain informed on regulatory changes, and who look for opportunities as the business landscape evolves, will have the best chance for guiding their labs to success.
That would certainly be true if CMS is able to publish a final rule that shifts a large number of procedures away from inpatient care and categorizes them as outpatient procedures.
Clinical laboratories are advised to continue developing methods for making prices for procedures available to the general public
Even as an effective treatment for COVID-19 continues to elude federal healthcare agencies, Medicare officials are pressing ahead with efforts to bring about transparency in hospital healthcare pricing, including clinical laboratory procedures and prescription drugs costs.
In FY 2021 Proposed Rule CMS-1735-P, titled, “Medicare Program; Hospital Inpatient Prospective Payment Systems for Acute Care Hospitals and the Long-Term Care Hospital Prospective Payment System and Proposed Policy Changes and Fiscal Year 2021 Rates; Quality Reporting and Medicare and Medicaid Promoting Interoperability Programs Requirements for Eligible Hospitals and Critical Access Hospitals,” the Centers for Medicare and Medicaid Services (CMS) proposes to “revise the Medicare hospital inpatient prospective payment systems (IPPS) for operating and capital-related costs of acute care hospitals to implement changes arising from our continuing experience with these systems for FY 2021 and to implement certain recent legislation.”
The proposed rule suggests a 1.6% increase (about $2 billion) in reimbursement for hospital inpatient services for 2021, but also eludes to the possibility of payer negotiated rates being used to determine future payment to hospitals.
In its analysis of the proposed rule, Modern Healthcare noted that CMS is “continuing its price transparency push, to the chagrin of some providers.”
However, the provisions in the proposed rule do, according to the CMS news release, advance several presidential executive orders, including:
Controversial Use of Payer Data for Future Medicare Rates
This latest CMS proposed rule (comments period ended July 10) moves forward “controversial price transparency” and has a new element of possible leverage of reported information for future Medicare payment rates, Healthcare Dive reported.
The 1,602-page proposed rule (CMS-1735-P) calls for these requirements in hospital Medicare cost reports:
Median payer-specific negotiated inpatient services;
Inclusion of rates for Medicare Advantage plans and other third party plans;
“In addition, the agency is requesting information regarding the potential use of these data to set relative Medicare payment rates for hospital procedures,” the CMS news release states.
Thus, under the proposed rule, the nation’s 3,200 acute care hospitals and 360 long-term care hospitals would need to start reporting requested data for discharges effective Oct. 1, 2020, a CMS fact sheet explained.
In the news release following the release of the proposed rule, CMS Administrator Seema Verma had a positive spin. “Today’s payment rate announcement focuses on what matters most to help hospitals conduct their business and receive stable and consistent payment.”
However, the American Hospital Association (AHA) articulated a different view, even calling the requirement for hospitals to report private terms “unlawful.”
“We are very disappointed that CMS continues down the unlawful path of requiring hospitals to disclose privately negotiated contract terms,” AHA Executive Vice President Tom Nickels (above) said in a statement, adding, “The disclosure of privately negotiated rates will not further CMS’ goal of paying market rates that reflect the cost of delivering care. These rates take into account any number of unique circumstances between a private payer and a hospital and simply are not relevant for fixing Fee-for-Service Medicare reimbursement.” (Photo copyright: American Hospital Association.)
AHA and other organizations attempted to block a price transparency final rule last year in a lawsuit filed against the U.S. Department of Health and Human Services (HHS), which oversees CMS, Dark Daily reported.
During in-court testimony, provider representatives declared that revealing rates they negotiate with payers violates First Amendment rights, Becker’s Hospital Review reported.
Officials for the federal government pushed back telling the federal judge that they can indeed require hospitals to publish negotiated rates. Hospital chargemasters, they added, don’t tell the full story, since consumers don’t pay those rates, Modern Healthcare reported.
In addition to the increase in inpatient payments and price transparency next steps, the recent CMS proposed rule also includes a new hospital payment category for chimeric antigen receptor (CAR) T-cell therapy. The technique uses a patient’s own genetically-modified immune cells to treat some cancers, as an alternative to chemotherapy and other treatment covered by IPPS, CMS said in the news release.
The agency also expressed intent to remove payment barriers to new antimicrobials approved by the FDA’s Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD pathway). “The LPAD pathway encourages the development of safe and effective drug products that address unmet needs of patients with serious bacterial and fungal infections,” the CMS fact sheet states.
Clinical laboratories are gateways to healthcare. For hospital lab leaders, the notion of making tests prices easily accessible to patients and consumers will soon no longer be a nice idea—but a legal requirement.
Therefore, clinical laboratory leaders are advised to stay abreast of price transparency regulations and continue to prepare for sharing test prices and information with patients and the general public in ways that fulfill federal requirements.