Though response was limited in Dark Daily’s poll, the message from respondents was overwhelmingly negative on LDT regulation by the FDA
Most respondents to a recent industry survey said that should Food and Drug Administration (FDA) approval be required in the future for laboratory developed tests (LDTs), innovation will suffer.
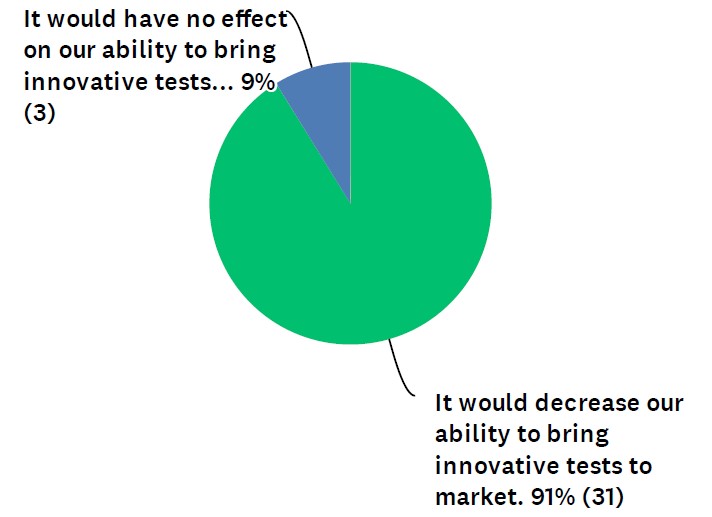
In the survey, which was conducted by Dark Daily, 91% of respondents said FDA pre-market approval of LDTs would decrease clinical laboratories’ ability to bring innovative tests to market. (See Figure 1.) The other 9% felt it would have no effect on innovation. Zero of the respondents said FDA involvement would increase innovative tests.
Figure 1: “How might a requirement of FDA pre-market approval impact the ability of your lab (or a lab you work for) to bring innovative tests to market?“
“Development of LDT tests has been the mission of most of the labs, and it meets the need for patient care,” noted one respondent in the survey. “Moving LDTs under FDA will create more obstacles for labs to offer the tests.”
To be fair, the survey had limited responses—34 in total. The poll went out to thousands of Dark Daily readers. We found that response rate surprising given how many labs will be affected if the VALID Act becomes law.
The VALID Act is a bipartisan bill that proposes FDA oversight of laboratory developed tests. The bill continues to make its way through the U.S. Senate and the House of Representatives.
A counterproposal called the Verified Innovative Testing in American Laboratories Act (VITAL Act) is also before Congress but has less momentum behind it. The VITAL Act seeks to keep LDTs under CLIA while also calling for reforms to account for modern lab tests.
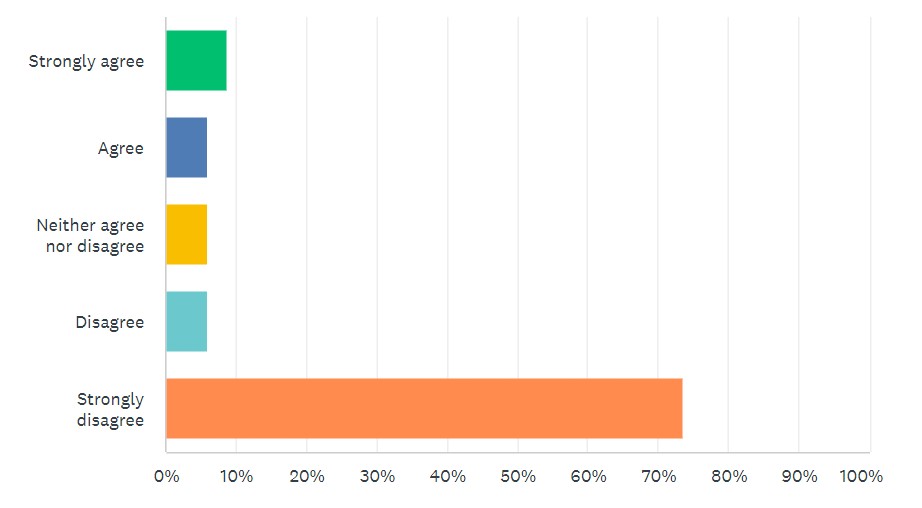
Looking at survey results, 80% “strongly disagreed” or “disagreed” that new LDT requirements, such as those found in the VALID Act, are needed. (See Figure 2.)
Figure 2: “FDA pre-market approval of LDTs should be required, as proposed in the VALID Act.”
By comparison, 65% “strongly agreed” or “agreed” with modernizing CLIA requirements for LDTs, as called for in the VITAL Act.
Those numbers shifted somewhat depending on the lab setting of the respondent. For example, just looking at commercial labs, opposition to the VALID Act remained similar, but support for modernizing CLIA jumped up to 88%. When looking at just hospitals, independent labs, and academic labs, numbers for both topics remained consistent.
When filtering the answers, the number of lab employees in a setting had little effect on survey results.
Political Battle Continues Over Laboratory Developed Tests
Clinical laboratory industry groups and others have been amassing to oppose or support the VALID Act. For example, the Advanced Medical Technology Association and The Pew Charitable Trusts are behind the bill.
However, the American Association for Clinical Chemistry, Association for Molecular Pathology, and new Coalition for Innovative Laboratory Testing are against the VALID Act.
Survey respondents can give their opinions about the proposed VALID and VITAL acts
Two bills are pending in Congress, and each is written to change the current regulatory scheme for laboratory-developed tests (LDTs) and in vitro clinical tests (IVCTs). The bills go by the acronyms of the VALID Act and VITAL Act. Many clinical laboratories offering LDTs today may be unaware of the details within each bill as currently written.
That existing regulatory arrangement will change if one of the two pending bills in Congress were to pass and be signed into law. That proposal is known as the Verifying Accurate Leading-Edge IVCT Development Act, or VALID Act. It is a bipartisan, 245-page bill that proposes FDA oversight of LDTs and is making its way through both the Senate and the House of Representatives.
A smaller, seven-page counterproposal is also before the Senate called the Verified Innovative Testing in American Laboratories Act, or VITAL Act. The VITAL Act would keep LDTs under CLIA but mandate updates to CLIA’s rules to account for modern tests.
Readers: Are you in favor of more or less regulation of LDTs? Take this quick survey and let us know what you think. Dark Daily wants to know your thoughts about LDT oversight. Click here to take our six-question survey. Results of this survey will be reported in a coming Dark Daily e-briefing.
Alert pathologists and clinical laboratory managers know that behind every bill proposed in Congress is a party with a vested interest that brought the issue to a senator or representative. Once enacted into law, a new bill changes the status quo, generally to the benefit of the private interests that requested that bill. This is true of both the VALID Act and the VITAL Act.
The table at the bottom of this briefing compares the provisions of each act and is current as of March 28.
Who Opposes VALID Act?
The VALID Act is garnering more attention than the VITAL Act.
On March 22, the American Association for Clinical Chemistry (AACC) sent out an email message urging its members to oppose the VALID Act.
“Let your legislators know that that if VALID becomes law, your institution and other hospitals and small commercial laboratories could be forced to stop providing LDTs,” wrote Patricia Jones, PhD, DABCC, FACB, Chair of AACC’s Policy and External Affairs Core Committee. The AACC has long criticized the VALID Act..
On the other side of the debate, Philadelphia-based The Pew Charitable Trusts, a nonprofit that in part analyzes publics policy, has come out in support of the VALID Act’s proposed requirements.
Two bills are pending in Congress about the future of LDT regulation.
“Although the [current] LDT regulatory process offers labs significant flexibility and enables a more rapid response to public health needs when no FDA-cleared or -approved test exists, the relative lack of oversight for LDTs puts the health of patients at risk,” Pew wrote in an October 2021 report on LDTs.
The Advanced Medical Technology Association also supports the VALID Act, as do many manufacturers of in vitro test kits and large commercial labs. Proponents also believe FDA regulation is needed for IVCTs because they are similar to medical devices and bring with them patient safety concerns.
The American Clinical Laboratory Association and the National Independent Laboratory Association (NILA) have not taken formal positions on the VALID Act.
Congress Could Roll VALID Act into MDUFA Vote to Win Passage
There may be an effort to attach the VALID Act to the authorization vote for the Medical Device User Fee Agreement V (MDUFA), according to a February health legislation alert from law firm Akin Gump Strauss Hauer & Feld based in Washington.
MDUFA funding provides resources to the FDA’s medical device review program. Congress is set to receive final MDUFA V recommendations in April.
Nineteen healthcare and lab industry groups, including the American Medical Association, AACC, AMP, and NILA, sent a joint letter to four Congress members on Feb. 23 requesting they deliberate the VALID Act separately and not as part of MDUFA.
Again, please complete this survey and tell us what you think about FDA regulation of LDTs, as defined in the VALID Act, compared to continuing LDT oversight via a modernized CLIA in the VITAL Act.
—Scott Wallask
Comparison of VALID Act and VITAL Act
VALID Act
VITAL Act
Full act name
Verifying Accurate Leading-Edge IVCT Development Act
Verified Innovative Testing in American Laboratories Act
Bill numbers
House Bill H.R.4128 Senate Bill S.2209
Senate Bill S.1666
Sponsors
Sen. Michael Bennet (D-CO) , Sen. Mike Braun (R-IN), Rep. Larry Bucshon, MD (R-IN), Sen. Richard Burr (R-NC), and Rep. Diana DeGette (D-CO)
Sen. Rand Paul (R-KY)
Provisions
Developers shall apply for premarket approval of IVCTs if there is insufficient evidence of analytical validity or clinical validity or if it’s reasonably possible an IVCT will cause serious adverse health effects.
Applications shall include a summary of test data and scientific evidence to support analytical and clinical validity of the test.
Through a technology certification, developers can submit an IVCT to the FDA for review, and if granted, the certification allows them to develop similar tests without going back for review each time.
The FDA must establish a program for rapid review of breakthrough IVCTs that provide effective treatment of life-threatening diseases
The federal government should work to ensure that regulatory oversight of laboratory tests does not limit patient access, impede innovation, or limit a test’s sustainability as a result of being unduly burdensome or beyond the fiscal capacity of the laboratory to reasonably validate and perform.
No aspects of LTDs shall be regulated under the FDA.
No later than 180 days after enactment of the bill, the secretary of health and human services shall report to the Senate’s Committee on Health, Education, Labor, and Pensions about recommendations to update clinical lab regulations and provide an assessment of LDT use during the 2020 pandemic response.
Exemptions
IVCTs being marketed before the VALID Act goes into effect
Low-risk tests
IVCTs that are granted emergency use
No new exemptions
Review timelines
The FDA shall make a decision no later than 90 days after an application is submitted.
No new requirements noted.
Sources: VALID Act and VITAL Act bills. Information is current as of March 28, 2022.
Though experts say an antigen test is not as accurate as PCR tests, its low cost, ease of use, and widespread availability make it a boon for clinical labs performing COVID-19 testing
As former FDA commissioner Scott Gottlieb, MD, explained on Face the Nation, “this kind of technology is a real game changer … it’s a very rapid test that could be used in a doctor’s office. Doctors now have about forty thousand of these Sofia machines already installed in their offices … you do a simple nasal swab and the test itself scans for the antigens that the virus produces.
“The test is about 85% sensitive. So, let’s say a hundred people come into a doctor’s office who have COVID-19, eighty-five of them are going to be able to be tested positive with this test very quickly. It’s a cheap test. It’ll probably be about five dollars a test and you can get a result within five minutes … you’re getting a very fast result and you can start to take action immediately.
“The company itself said that they’re going to be able to produce about two hundred thousand of these tests starting right away. But in several weeks, they’ll be able to produce up to 1.5 million a week. So, this dramatically expands our testing capacity as long as doctors are able to run these tests in their offices.”
In an interview on CBS’ Face the Nation, former FDA Commissioner Scott Gottlieb, MD (above), said, “These antigen-based tests aren’t as reliable, meaning they’re not as sensitive. So, they’re going to miss some patients who have COVID. But in the hands of a doctor who already has a high index of suspicion that the patient may have the disease … they allow you to dramatically expand testing. And they’re very cheap. They’re very easy to perform. And again, most doctors have these machines already in their offices.” (Photo copyright: US Food and Drug Administration.)
Other LDTs That Have Received EUAs
Here’s a look at other laboratory-developed tests from major manufacturers that have received emergency-use authorizations from the FDA:
This test is designed for use with Abbott’s m2000 RealTime system, which is installed in about 200 US medical laboratories, the company says. It can run up to 470 patient samples in 24 hours. As of a May 11 statement, the company said it had shipped more than two million tests in the US.
This test is designed for use with Abbott’s Alinity m system, which the company describes as its “most advanced laboratory molecular instrument,” with the ability to run up to 1,080 tests in 24 hours, according to a press release.
This is a rapid test designed for use with the ID Now system, a compact portable instrument for point-of-care settings such as urgent care clinics. As of May 11, Abbott said it had shipped more than 1.7 million tests in the US, and that it planned to increase manufacturing capacity to two million tests per month.
However, the test has encountered some stumbling blocks. On May 14, the FDA issued an alert stating that the ID Now COVID-19 test could produce inaccurate negative results. This came after researchers at NYU Langone Health, Northwell Health, and Cleveland Clinic reported problems with the test, according to MedTech Dive. Abbott issued a statement suggesting that the problems were due to improper sample collection and handling, however, the FDA said that Abbott had agreed to conduct post-market studies to identify the cause of the false negatives and suggest remedial actions.
This is a qualitative test designed to detect the presence of IgG antibodies following a SARS-CoV-2 infection. The FDA authorized use of the assay on Abbott’s Architect i2000SR system in April, and then followed up with a May 11 EUA for its use on the Alinity i system. In a statement, Abbott said it planned to ship 30 million tests globally starting in May.
In a March statement, the FDA touted this as the first point-of-care COVID-19 test to receive an EUA. The company estimates the detection time as approximately 45 minutes. It is designed for use with Cepheid’s GeneXpert Dx diagnostic software and GeneXpert Infinity systems, which have nearly 5,000 US installations, according to a Cepheid statement.
This test runs on Hologic’s Panther system, which, according to a Hologic press release, can provide results in about three hours and run more than 1,000 tests per day. The company claims that more than 1,000 Panther systems are installed in US labs, and that it expects to produce an average of one million tests per week.
Ortho’s antibody test is designed for use with its VITROS XT 7600, 3600, 5600, and ECi/ECiQ immunodiagnostic systems, which, the company says are installed in more than 1,000 US labs. The Total Reagent Pack is a qualitative test that detects the presence of all antibodies against SARS-CoV-2.
On April 24, Ortho announced it had received another FDA EUA, this one for its Anti-SARS-CoV-2 IgG test, which detects the presence of IgG antibodies. In a statement, the company said it expects to produce “several million” IgG tests per month.
This test is designed for use with Roche’s cobas 6800 and 8800 systems. The 6800 can process up to 384 results in an eight-hour shift, Roche says, compared with 1,056 results for the 8800 model. The company says results are available in about 3.5 hours. In a statement, Roche said it planned to ship 400,000 tests per week.
Roche describes this as a qualitative antibody test that can be used on cobas e series immunoassay analyzers. Testing time is 18 minutes. As of May 19, the test was live at more than 20 US labs, “with plans in the next several weeks to increase to more than 200 commercial and hospital lab sites with the ability to perform millions of tests per week,” the company stated in a press release.
It’s likely the FDA will continue to issue emergency-use authorizations as the agency receives more applications from IVD manufacturers.
By taking early measures to combat the spread, the country had a medical laboratory test for COVID-19 available as early as Jan. 24, and was able to focus medical laboratory testing on the most at-risk individuals
With the Coronavirus disease 2019 (COVID-19) outbreak dominating headlines and medical laboratories under growing pressure to increase testing capacity, Taiwan’s rapid response to the pandemic could provide a critical model for other countries to follow.
Given its proximity to mainland China—just 81 miles—and the large number of individuals who frequently travel back and forth between the countries, Taiwan was at risk of having the second-highest number of imported COVID-19 cases, according to a model developed by researchers at Johns Hopkins University and the University of New South Wales Sydney. News reports indicate that, each year, about 60,000 flights carry 10 million passengers between Taiwan and China.
Data from Taiwan’s Centers for Disease Control (CDC) and Central Epidemic Command Center (CECC) indicate that the country has managed to contain the outbreak thanks to these aggressive actions.
As of March 19, Taiwan’s CECC reported a total of 108 laboratory-confirmed COVID-19 infections. That compares with 81,155 in China, 41,035 in Italy, and 10,755 in the US, according to data compiled by the Center for Systems Science and Engineering at Johns Hopkins University. When the World Health Organization (WHO) reports on the number of COVID-19 cases by country, it includes the number of COVID-19 cases from Taiwan under the totals for the People’s Republic of China. WHO made this decision several years ago, under pressure by China to not recognize Taiwan as an independent nation.
The World
Population Review website says Taiwan’s population is about 23.8 million.
But its infection rate is low even on a per capita basis: Approximately 45
infections per million population, compared with 6,784 in Italy, 564 in China,
and 326 per million in the US.
The JAMA authors noted that Taiwan was prepared for
an outbreak after its experience with the severe
acute respiratory syndrome (SARS) pandemic in 2003, which also originated
in China.
Timeline of COVID-19 Outbreak at the Earliest Stages
Taiwan apparently learned a lesson about preparedness from
the SARS outbreak the rest of the world did not and that enabled the tiny
nation to respond immediately to the novel Coronavirus threat.
The country’s efforts began on Dec. 31 with inspections of
flight arrivals from Wuhan. “When there were only a very few cases [of
COVID-19] reported in China, [Taiwanese health authorities] already went onto
every airplane that came from Wuhan,” C. Jason Wang,
MD, PhD, an Associate Professor of Pediatrics and Director of the Center for Policy, Outcomes, and
Prevention at Stanford University and lead author of the JAMA
report, told Vox.
“Health officials came on the airplane and checked people for symptoms,” he
added.
Travelers who had recently visited Wuhan and displayed
symptoms of pneumonia were quarantined at home for 14 days. Taiwan’s
CDC reported that quarantined individuals were being tested for the
2019-nCoV coronavirus (later renamed to SARS-CoV-2)
soon after it was identified. The CECC, activated in January to coordinate the
government’s response, reported the first confirmed imported case on Jan. 21.
On Jan. 24, their
CDC announced that testing for the virus was being performed at the CDC and
eight designated hospitals. Testing included samples from physicians around the
country. As of Feb. 17, daily testing capacity was about 1,300 samples, the JAMA
authors reported.
Wang told Vox that aggressive measures to identify
and isolate at-risk individuals at the earliest stages reduced the volume of clinical
laboratory tests that had to be performed. “Here in the US and elsewhere, we’re
now seeing community spread,” he said. “It’s probably been here for a while.
And so now we’re trying to see, ‘Oh, how many people should we test?’ Then, you
really need to have a very large capacity in the beginning.”
“I think the US has enormous capacity that’s currently not being used,” C. Jason Wang, MD, PhD (above), Associate Professor of Pediatrics and Director of the Center for Policy, Outcomes, and Prevention at Stanford University and lead author of the JAMA report, told Vox. “We have big tech companies that really could do a lot, right? We ought to get the big companies together. Get the governors together, get the federal government agencies to work with each other, and try to find innovative ways to think about how to best do this. We’ve got the smartest people here in the US because they come from everywhere. But right now, those are untapped resources. They’re not working together. And the federal government, the agencies, they need to collaborate a little more closely.” (Photo copyright: Stanford University.)
More Actions by Authorities
The JAMA report supplementary materials notes a total of 124 actions taken by Taiwanese authorities between Jan. 20 and Feb. 24 to contain the outbreak. In addition to the border inspections, quarantines and testing, they included integration of data between the country’s National Health Insurance Administration and National Immigration Agency, so authorities, and later hospitals, could identify any patient who had recently traveled to China, Hong Kong, or Macau.
The steps also included:
An escalating series of travel restrictions,
eventually including suspension of most passenger flights from Taiwan to China,
as well as a suspension of tours to Hong Kong or Macau.
Use of government-issued cell phones to monitor
quarantined individuals.
Fines for individuals breaking the 14-day home
quarantine.
Fines for incoming travelers who failed to
provide accurate health information.
Fines for disseminating false information or
rumors about the epidemic.
Fines and jail sentences for profiteering on disease-prevention
products.
Designation of military camps and other
government facilities for quarantine.
Nationwide disinfection of universities,
colleges, and public spaces around schools.
The government also took aggressive action to ensure
adequate supplies of surgical masks, including stepped-up manufacturing, export
bans, price limits, and a limit of one to three masks per purchase.
The JAMA authors noted that government officials issued daily press briefings to educate the public about the outbreak. Communication efforts also included public service announcements by Taiwan Vice President Chen Chien-jen, a trained epidemiologist.
A poll taken in Taiwan on Feb. 17 and 18 indicated high approval ratings for officials’ response to the crisis.
The JAMA authors also noted some “challenges” in the
government’s response. For example, most real-time public communication was in
Mandarin Chinese and sign language, leaving out non-Taiwanese citizens in the
country. And the cruise ship Diamond Princess, later found to have infections
on board, was allowed to dock near Taipei and disembark passengers. There are
also questions about whether similar policies can be sustained through the end
of a pandemic.
Still, “well-trained and experienced teams of officials were
quick to recognize the crisis and activated emergency management structures to
address the emerging outbreak,” the JAMA authors wrote. “Taiwan is an
example of how a society can respond quickly to a crisis and protect the
interests of its citizens.”
One noteworthy difference in the speedy response to
recognition of a novel coronavirus in Taiwan, compared to recognition of the
same novel coronavirus in the United States, was the fast availability of
clinical laboratory tests for COVID-19 in Taiwan.
Pathologists and clinical laboratory professionals here in
the US are frustrated that their skills and talents at developing and
validating new assays on an accelerated timeline were not acknowledged and
leveraged by government officials as they decided how to respond to the
emergence of the novel coronavirus now called SARS-CoV-2.
Scientist described the speed at which SARS-CoV-2’s full sequence of genetic material was made public as ‘unprecedented’ and medical labs are rushing to validate tests for this new disease
In the United States, headlines scream about the lack of
testing for the novel Coronavirus
disease 2019 (COVID-19). News reporters ask daily why it is taking so long
for the US healthcare system to begin testing large numbers of patients for
SARS-CoV-2, the virus that causes COVID-19. Yet, pathologists
and clinical
laboratory scientists know that new technologies for gene sequencing
and diagnostic testing are helping public health laboratories bring up tests
for a previously unknown new disease faster than at any time in the past.
At the center of the effort to develop accurate new assays
to detect SARS-CoV-2 and help diagnose cases of the COVID-19 disease are medical laboratory
scientists working in public health
laboratories, in academic medical centers, and in research labs across the
United States. Their collective efforts are producing results on a faster
timeline than in any previous discovery of a new infectious disease.
For example, during the severe
acute respiratory syndrome (SARS) outbreak in 2003, five months passed
between the first recognized case of the disease in China and when a team of
Canadian scientists cracked the genetic code of the virus, which was needed to
definitively diagnose SARS patients, ABC
News reported.
In contrast, Chinese scientists sequenced this year’s
coronavirus (originally named 2019-nCoV) and made it available on Jan. 10,
2020, just weeks after public health officials in Wuhan, China, reported the
first case of pneumonia from the unknown virus to the World Health Organization
(WHO), STAT
reported.
Increases in sequencing speed enabled biotechnology
companies to quickly create synthetic copies of the virus needed for research. Roughly
two weeks later, scientists completed sequencing nearly two dozen more samples
from different patients diagnosed with COVID-19.
Molecular biologist Kristian Andersen, PhD (above right, with graduate students who helped sequence the Zika virus), an Associate Professor in the Department of Immunology and Microbiology at Scripps Research in California and Director of Infectious Disease Genomics at Scripps’ Translational Research Institute, worked on the team that sequenced the Ebola genome during the 2014 outbreak. He told STAT that the pace of sequencing of the SARS-CoV-2 coronavirus is “unprecedented.” (Photo copyright: Scripps Research.)
Lower Sequencing Costs Speed COVID-19 Diagnostics Research
Additionally, a significant decline in the cost of genetic synthesis is playing an equally important role in helping scientists slow the spread of COVID-19.In its coverage of the SARS-CoV-2 outbreak, The Verge noted that two decades ago “it cost $10 to create a synthetic copy of one single nucleotide, the building block of genetic material. Now, it’s under 10 cents.” Since the coronavirus gene is about 30,000 nucleotides long, that price reduction is significant.
Faster sequencing and cheaper access to synthetic copies is
contributing to the development of diagnostic tests for COVID-19, an important
step in slowing the disease.
“This continues to be an evolving situation and the ability to distribute this diagnostic test to qualified medical laboratories is a critical step forward in protecting the public health,” FDA Commissioner Stephen M. Hahn, MD, said in an FDA statement.
However, the Washington Post soon reported that the government-created coronavirus test kits contained a “faulty component,” which as of February 25 had limited testing in the US to only 426 people, not including passengers who returned to the US on evacuation flights. The Post noted that the nation’s public health laboratories took “the unusual step of appealing to the FDA for permission to develop and use their own [laboratory-developed] tests” for the coronavirus.
“This is an extraordinary request, but this is an extraordinary time,” Scott Becker,
Parallel efforts to develop and validate tests for COVID-19
are happening at the clinical laboratories of academic medical centers and in a
number of commercial laboratory companies. As these labs show their tests meet
FDA criteria, they become available for use by physicians and other healthcare
providers.
Dark Daily’s sister publication, The Dark Report just published an intelligence briefing about the urgent effort at the clinical laboratory of Northwell Health to develop both a manual COVID-19 assay and a test that can be run on the automated analyzers already in use in the labs at Northwell Health’s 23 hospitals. (See TDR, “Northwell Lab Team Validates COVID-19 Test on Fast Timeline,” March 9, 2020.)
Following the FDA’s March 13 EUA for the Thermo Fisher test,
Hahn said, “We have been engaging with test developers and encouraging them to
come to the FDA and work with us. Since the beginning of this outbreak, more
than 80 test developers have sought our assistance with development and
validation of tests they plan to bring through the Emergency Use Authorization
process. Additionally,” he continued, “more than 30 laboratories have notified
us they are testing or intend to begin testing soon under our new policy for
laboratory-developed tests for this emergency. The number of products in the
pipeline reflects the significant role diagnostics play in this outbreak and
the large number of organizations we are working with to bring tests to
market.”
Pharma Company Uses Sequencing Data to Develop Vaccine in
Record Time
Even as clinical laboratories work to develop and validate diagnostic tests for COVID-19, drug manufacturers are moving rapidly to develop a COVID-19 vaccine. In February, Massachusetts-based biotechnology company Moderna Therapeutics (NASDAQ:MRNA) announced it had shipped the first vials of its potential coronavirus vaccine (mRNA-1273) to the National Institute of Allergy and Infectious Disease (NIAID) for use in a Phase One clinical trial.
“The collaboration across Moderna, with NIAID, and with CEPI [Coalition for Epidemic Preparedness Innovations] has allowed us to deliver a clinical batch in 42 days from sequence identification,” Juan Andres, Chief Technical Operations and Quality Officer at Moderna, stated in a news release.
The Wall Street Journal (WSJ) reported that NIAID expects to start a clinical trial of about 20 to 25 healthy volunteers by the end of April, with results available as early as July or August.
“Going into a Phase One trial within three months of getting the sequence is unquestionably the world indoor record,” NIAID Director Anthony Fauci, MD, told the WSJ. “Nothing has ever gone that fast.”
There are no guarantees that Moderna’s coronavirus vaccine
will work. Furthermore, it will require further studies and regulatory
clearances that could delay widespread distribution until next year.
Nonetheless, Fauci told the WSJ, “The only way you
can completely suppress an emerging infectious disease is with a vaccine. If
you want to really get it quickly, you’re using technologies that are not as
time-honored as the standard, what I call antiquated, way of doing it.”
In many ways, the news media has overlooked all the important
differences in how fast useful diagnostic and therapeutic solutions for
COVID-19 are moving from research settings into clinical use, when compared to
early episodes of the emergence of a new infectious disease, such as SARS in
2003.
The story the American public has yet to learn is how new
genetic sequencing technologies, improved diagnostic methods, and enhanced
informatics capabilities are being used by researchers, pathologists, and
clinical laboratory professionals to understand this new disease and give
healthcare professionals the tools they need to diagnose, treat, and monitor
patients with COVID-19.