This is important for clinical laboratory leaders to watch, because medical labs often interface with hospital EHRs to exchange vital patient data, a key component of complying with Medicare’s EHR incentive programs. If claims of interoperability are shown to be false, could labs engaged with those hospital systems under scrutiny be drawn into the DOJ’s investigations?
Violating the False Claims Act
In May, Coffey Health System (CHS), which includes Coffey County Hospital, a 25-bed critical access hospital located in Burlington, Kan., agreed to pay the US government a total of $250,000 to settle a claim that it violated the False Claims Act.
CHS’ former CIO filed the qui tam (aka, whistleblower) lawsuit, which allows individuals to sue on behalf of the government and share in monetary recovery. He alleged that CHS provided false information to the government about being in compliance with security standards to receive incentive payments under the EHR Incentive Program.
According to a DOJ press release, “the United States alleged that Coffey Health System falsely attested that it conducted and/or reviewed security risk analyses in accordance with requirements under a federal incentive program for the reporting periods of 2012 and 2013. The government contended that the hospital submitted false claims to the Medicare and Medicaid Programs pursuant the Electronic Health Records (EHR) Incentive Program.”
“Medicare and Medicaid beneficiaries expect that providers ensure the accuracy and security of their electronic health records,” said Stephen McAllister (above), United States Attorney for the District of Kansas, in the DOJ press release. “This office remains committed to protecting the federal health programs and to hold accountable those whose conduct results in improper payments.” (Photo copyright: US Department of Justice.)
The Recovery Act allocated $25 billion to incentivize healthcare professionals and facilities to adopt and demonstrate meaningful use (MU) of electronic health records by January 1, 2014. The federal Centers for Medicare and Medicaid Services (CMS) released the incentive funds when providers attested to accomplishing specific goals set by the program.
The website of the Office of the National Coordinator for Health Information Technology (ONC), HealthIt.gov, defines “meaningful use” as the use of digital medical and health records to:
Improve quality, safety, efficiency, and reduce
health disparities;
Engage patients and their families;
Improve care coordination and population and
public health; and
Maintain privacy and security of patient health
information.
The purpose of the HITECH Act was to address privacy and security concerns linked to electronic storage and transference of protected health information (PHI). HITECH encourages healthcare organizations to update their health records and record systems, and it offers financial incentives to institutions that are in compliance with the requirements of the program.
When eligible professionals or eligible hospitals attest to being in compliance with Medicare’s EHR incentive program requirements, they can file claims for federal funds, which are paid and audited by the Department of Health and Human Services (HHS) through Medicare and Medicaid.
Institutions receiving funds must demonstrate meaningful use
of EHR records or risk potential penalties, including the delay or cancellation
of future payments and full reimbursement of payments already received. In
addition, false statements submitted in filed documents are subject to criminal
laws and civil penalties at both the state and federal levels.
EHR Developers Under Scrutiny by DOJ
EHR vendors also have been investigated and ordered to make
restitutions by the DOJ.
In February, Greenway Health, a Tampa-based EHR developer, agree to pay $57.25 million to resolve allegations related to the False Claims Act. In this case, the government contended that Greenway obtained certification for its “Prime Suite” EHR even though the technology did not meet the requirements for meaningful use.
And EHR vendor eClinicalWorks paid the government $155 million to settle allegations under the False Claims Act. The government maintained that eClinicalWorks misrepresented the capabilities of their software and provided $392,000 in kickbacks to customers who promoted its product.
Legal cases such as these demonstrate that the DOJ will
pursue both vendors and healthcare organizations that misrepresent their
products or falsely attest to interoperability under the terms laid out by
Medicare’s EHR Incentive Program.
Clinical laboratory leaders and pathology groups should carefully
study these cases. This knowledge may be helpful when they are asked to create
and maintain interfaces to exchange patient data with client EHRs.
Despite the widespread adoption of electronic health record (EHR) systems and billions in government incentives, lack of interoperability still blocks potential benefits of digital health records, causing frustration among physicians, medical labs, and patients
Clinical laboratories and anatomic pathology groups understand the complexity of today’s electronic health record (EHR) systems. The ability to easily and securely transmit pathology test results and other diagnostic information among multiple providers was the entire point of shifting the nation’s healthcare industry from paper-based to digital health records. However, despite recent advances, true interoperability between disparate health networks remains elusive.
One major reason for the current situation is that multi-hospital health systems and health networks still use EHR systems from different vendors. This fact is well-known to the nation’s medical laboratories because they must spend money and resources to maintain electronic lab test ordering and resulting interfaces with all of these different EHRs.
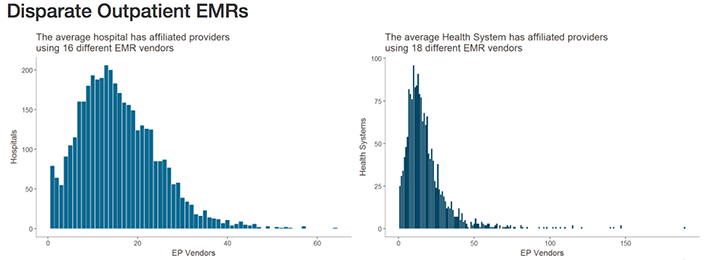
Healthcare IT News highlighted the scale of this problem in recent coverage. Citing data from the Healthcare Information and Management Systems Society (HIMSS) Logic database, they note that—when taking into account affiliated providers—the typical health network engages with as many as 18 different electronic medical record (EMR) vendors. Similarly, hospitals may be engaging with as many as 16 different EMR vendors.
The graphics above illustrates why interoperability is the most important hurdle facing healthcare today. Although the shift to digital is well underway, medical laboratories, physicians, and patients still struggle to communicate data between providers and access it in a universal or centralized manner. (Images copyright: Healthcare IT News.)
The lack of interoperability forces healthcare and diagnostics facilities to develop workarounds for locating, transmitting, receiving, and analyzing data. This simply compounds the problem.
Pressure from Technology Giants Fuels Push for Interoperability
According to HITECH Answers, the Centers for Medicare and Medicaid Services (CMS) has paid out more than $38-billion in EHR Incentive Program payments since April 2018.
Experts, however, point out that government incentives are only one part of the pressure vendors are seeing to improve interoperability.
“There needs to be a regulatory push here to play referee and determine what standards will be necessary,” Blain Newton, Executive Vice President, HIMSS Analytics, told Healthcare IT News. “But the [EHR] vendors are going to have to do it because of consumer demand, as things like Apple Health Records gain traction.”
Another solution, according to TechTarget, involves developing application programming interfaces (APIs) that allow tech companies and EHR vendors to achieve better interoperability by linking information in a structured manner, facilitating secure data transmission, and powering the next generation of apps that will bring interoperability ever closer to a reality.
TechTarget reported on how University of Utah Hospital’s five hospital/12 community clinic health network, and Intermountain Healthcare, also in Utah, successfully used APIs to develop customized interfaces and apps to improve accessibility and interoperability with their Epic and Cerner EHR systems.
Diagnostic Opportunities for Clinical Laboratories
As consumers gain increased access to their data and healthcare providers harness the current generation of third-party tools to streamline EHR use, vendors will continue to feel pressure to make interoperability a native feature of their EHR systems and reduce the need to rely on HIT teams for customization.
For pathology groups, medical laboratories, and other diagnosticians who interact with EHR systems daily, the impact of interoperability is clear. With the help of tech companies, and a shift in focus from government incentives programs, improved interoperability might soon offer innovative new uses for PHI in diagnosing and treating disease, while further improving the efficiency of clinical laboratories that face tightening budgets, reduced reimbursements, and greater competition.
Softened FDA regulation of both clinical-decision-support and patient-decision-support software applications could present opportunities for clinical laboratory developers of such tools
Physician decision-support software utilizes medical laboratory test data as a significant part of a full dataset used to guide caregivers. Thus, if the FDA makes it easier for developers to get regulatory clearance for these types of products, that could positively impact medical labs’ ability to service their client physicians.
Additionally, clinical pathologists have unique training in diagnosing diseases and understanding the capabilities and limitations of medical laboratory tests in supporting how physicians diagnose disease and make treatment decisions. Thus, actions by the FDA to make it easier for developers of software algorithms that can incorporate clinical laboratory data and anatomic pathology images with the goal of improving diagnoses, decisions to treat, and monitoring of patients have the potential to bring great benefit to the nation’s medical laboratories.
FDA Clarifies Role in Regulating CDS/PDS Applications
The new guidelines clarified items specified in the 21st Century Cures Act, which was enacted by Congress in December of 2016. This Act authorized $6.3 billion in funding for the discovery, development, and delivery of advanced, state-of-the art medical cures.
“Today, we’re announcing three new guidances—two draft and one final—that address, in part, important provisions of the 21st Century Cures Act, that offer additional clarity about where the FDA sees its role in digital health, and importantly, where we don’t see a need for FDA involvement,” FDA commissioner Scott Gottlieb, MD, Commissioner of Food and Drugs, noted in a statement. “We’ve taken the instructions Congress gave us under the Cures Act and [we] are building on these provisions to make sure that we’re adopting the full spirit of the goals we were entrusted with by Congress.”
Helping Doctors’ Decision-Making
The first guideline concerns clinical decision support systems that are designed to help doctors make data-driven decisions about patient care. The new guidelines make it easier for software developers to get regulatory clearance, which, the FDA hopes, will spark innovation and makes regulation more efficient.
“CDS has many uses, including helping providers, and ultimately patients, identify the most appropriate treatment plan for their disease or condition,” Gottlieb said in the FDA’s statement. “For example, such software can include programs that compare patient-specific signs, symptoms, or results with available clinical guidelines to recommend diagnostic tests, investigations or therapy.
“This type of technology has the potential to enable providers and patients to fully leverage digital tools to improve decision making,” Gottlieb continued. “We want to encourage developers to create, adapt, and expand the functionalities of their software to aid providers in diagnosing and treating old and new medical maladies.”
Identifying Digital Health Applications That Receive/Don’t Receive FDA Oversight
The second guideline discusses and delineates which digital health applications are considered low risk and, thus, will not fall under FDA regulations.
Products that are not intended to be used for the diagnosis, cure, mitigation, prevention, or treatment of a condition will not be regulated by the FDA. These technologies are not considered medical devices and may include gadgets such as weight management and mindfulness tools. They can provide value to consumers and the healthcare industry while posing a low risk to patients.
“Similarly, the CDS draft guidance also proposes to not enforce regulatory requirements for lower-risk decision support software that’s intended to be used by patients or caregivers—known as patient-decision-support software (PDS)—when such software allows a patient or a caregiver to independently review the basis of the treatment recommendation,” Gottlieb noted in the statement.
Scott Gottlieb, MD (above), FDA Commissioner of Food and Drugs, noted in a statement, “We believe our proposals for regulating CDS and PDS not only fulfill the provisions of the Cures Act, but also strike the right balance between ensuring patient safety and promoting innovation. Clinical laboratories may find opportunities to work with CDS/PDS developers and support their client physicians. (Photo copyright: FDA.)
However, products that are intended to be used for the diagnosis, cure, mitigation, prevention, or treatment of a condition are considered medical devices and will fall under FDA regulations.
“The FDA will continue to enforce oversight of software programs that are intended to process or analyze medical images, signals from in vitro diagnostic devices, or patterns acquired from a processor like an electrocardiogram that use analytical functionalities to make treatment recommendations, as these remain medical devices under the Cures Act,” noted Gottlieb.
Items such as mobile apps that are utilized to maintain and encourage a healthy lifestyle are not deemed to be medical devices and will fall outside FDA regulations. The guidelines also defined that Office of the National Coordinator for Health Information Technology (ONC)-certified electronic health record (EHR) systems are not medical devices and, thus, will not be regulated by the FDA.
Software-as-a-Medical Device Gets FDA Oversight
The third guidance document deals with the assessment of the safety, performance, and effectiveness of Software as a Medical Device (SaMD).
“This final guidance provides globally recognized principles for analyzing and assessing SaMD, based on the overall risk of the product. The agency’s adoption of these principles provides us with an initial framework when further developing our own specific regulatory approaches and expectations for regulatory oversight and is another important piece in our overarching policy framework for digital health,” Gottlieb noted in the statement.
SaMD is defined by the International Medical Device Regulators Forum (IMDRF) as “software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”
Gottlieb noted that the three important guidance documents being issued would continue to expand the FDA’s efforts to encourage innovation in the ever-changing field of digital health. “Our aim is to provide more clarity on, and innovative changes to, our risk-based approach to digital health products, so that innovators know where they stand relative to the FDA’s regulatory framework. Our interpretation of the Cures Act is creating a bright line to define those areas where we do not require premarket review,” he concluded.
What remains to be seen is how the new FDA regulations will impact clinical laboratories and anatomic pathology groups. With the expanding interest in artificial intelligence (AI) and self-learning software systems, healthcare futurists are predicting a rosy future for informatics products that incorporate these technologies. Hopefully, with these new guidelines in place, innovative clinical laboratories will have the opportunity to develop new digital products for their clients.
Meaningful Use Stage 3 focuses on interoperability, which is good news for medical laboratories that must spend time and money to develop effective LIS-EHR interfaces
On December 15, 2015, the final rule for Stage 3 meaningful use (MU) went into effect. By now, pathologists and clinical laboratory managers and personnel are well-acquainted with the MU incentive program and the myriad of challenges it presents for almost everyone working in the healthcare sector.
That’s good news for providers struggling with EHR attestation. However, the struggle for clinical laboratories isn’t with attestation per se, it’s with interoperability between lab information systems (LIS) and physicians’ EHRs. (more…)
One new federal law forbids health IT vendors and providers from deliberately blocking information-sharing with competing EHR systems
Several years deep into its effort to get physicians and hospitals to use electronic health record (EHR) systems, the federal government has yet to come up with a way to improve interoperability—the ability of EHRs to interface and communicate with other systems.
Stage one and stage two Meaningful Use guidelines have failed to successfully address the barriers preventing interoperability. Of course, clinical laboratories and pathology groups encounter this problem daily. That’s because they must build interfaces between their laboratory information systems (LIS) and the EHRs of their client physicians. The cost of creating workable LIS-to-EHR interfaces continues to be a huge burden on medical laboratories and that is why they support improved interoperability. But labs also contribute to the lack of interoperability when they enact restrictions on how lab test data can be shared with other providers and competing labs who are serving the same physicians and patients. (more…)